Europe approves first gene therapy for treatment of haemophilia A
Posted: 30 August 2022 | Caroline Peachey (European Pharmaceutical Review) | No comments yet
European approval of BioMarin’s ROCTAVIAN (valoctocogene roxaparvovec) one-time gene therapy represents a breakthrough in the treatment of adults with severe haemophilia A.
The European Commission has granted conditional marketing authorization to BioMarin’s ROCTAVIAN™ (valoctocogene roxaparvovec) gene therapy for the treatment of severe haemophilia A in adult patients without a history of Factor VIII inhibitors and without detectable antibodies to adeno-associated virus serotype 5 (AAV5).
The EC also endorsed the European Medicines Agency’s (EMA’s) recommendation for Roctavian to maintain orphan drug designation, thereby granting a 10-year period of market exclusivity.
The EMA recommendation stated that, even in light of existing treatments, Roctavian may offer a “significant benefit” to those affected with severe Haemophilia A.
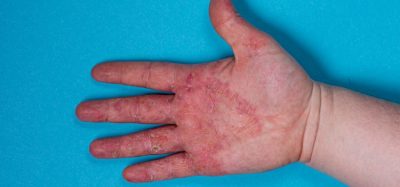
Haemophilia A is a rare debilitating disease affecting approximately 0.7 in 10,000 people in the European Union.
Patients with haemophilia A cannot produce factor VIII (an essential protein required for blood to clot and stop bleeding), which means they are more prone to bleeding and may suffer prolonged/life-threatening bleeds from modest injuries.
Roctavian — a one-time infusion gene therapy for haemophilia A — works by delivering a functional gene that is designed to enable the body to produce Factor VIII on its own without the need for continued haemophilia prophylaxis.
Current treatments require one or more injections on a weekly/monthly basis and are lifelong. Therefore, there is an unmet medical need for new therapeutic approaches that might free patients from frequent injections, EMA said.
“This approval in the EU represents a medical breakthrough in the treatment of patients with severe haemophilia A that expands the conversation between a patient and physician on treatment choices to now include a one-time infusion that protects from bleeds for several years,” stated Professor Johannes Oldenburg, Director of the Institute of Experimental Haematology and Transfusion Medicine and the Haemophilia Centre at the University Clinic in Bonn, Germany.
“Roctavian approval in Europe is a historic milestone in medicine and is built upon almost four decades of scientific discovery, innovation, and perseverance,” added Jean-Jacques Bienaimé, Chairman and CEO of BioMarin.
The EC based its decision on a significant body of data from the Roctavian clinical development programme, including two-year outcomes from the global GENEr8-1 Phase III study. Two years after the administration, efficacy data showed that the therapy significantly increased factor VIII activity levels in most patients, with bleeding rates reduced in 84% of cases. Data also included five and four years of follow-up from the 6e13 vg/kg and 4e13 vg/kg dose cohorts, respectively, in ongoing Phase I/II dose escalation study.
BioMarin has committed to continue working with the broader community and the EMA to monitor the long-term effects of treatment.
Severe haemophilia A is estimated to affect more than 20,000 adults across more than 70 countries in Europe, the Middle East, and Africa. BioMarin estimates there are 3,200 patients who will be indicated for Roctavian in the 24 countries covered by the EU approval.
BioMarin said it remains committed to bringing Roctavian to eligible patients with severe haemophilia A in the United States and is targeting a Biologics License Application (BLA) resubmission to the US Food and Drug Administration (FDA) for Roctavian by the end of September 2022.
BLA resubmissions are typically followed by a six-month review process. However, BioMarin said it anticipates an additional three-month review period based on the number of data read-outs that will emerge during the procedure.
Summer 2022 haemophilia trial update…
Related topics
Biologics, Drug Development, Drug Safety, Gene therapy, Orphan Drugs