Identification of protein biomarkers and the evaluation of changes in protein expression following drug treatment rely on both the generation of peptides from cellular proteins, and the acquisition and interpretation of spectra generated by tandem mass spectrometry (MS/MS). Acquisition of MS/MS spectra in a datadependent manner means that a significant number of the protein fragments (peptides) generated are never actually subjected to MS/MS1. Moreover, only a small proportion of acquired MS/MS spectra are ever interpreted, despite the large number of tools for the automated analysis of such data. Furthermore, many fragment ions are simply ignored during data analysis, in large part because automated search engines do not ‘look’ for all potential fragmentation products, and also because we simply still do not sufficiently understand the mechanisms of gas-phase peptide fragmentation to fully interpret the spectra (most likely a combination of the two). The end result is that even though proteome coverage is increasing in large-scale analyses, we are still a long way from the ideal of ‘complete’ proteome analysis.
FIGURE 1The b5 ion is generated by CID which terminates at the C-terminus in an oxazolone ring structure. N-terminal nucleophilic attack of the oxazolone ring results in formation of a macrocyclic intermediate that undergoes ring opening at various positions to generate sequence scrambled b5 ions.
Identification of protein biomarkers and the evaluation of changes in protein expression following drug treatment rely on both the generation of peptides from cellular proteins, and the acquisition and interpretation of spectra generated by tandem mass spectrometry (MS/MS). Acquisition of MS/MS spectra in a datadependent manner means that a significant number of the protein fragments (peptides) generated are never actually subjected to MS/MS1. Moreover, only a small proportion of acquired MS/MS spectra are ever interpreted, despite the large number of tools for the automated analysis of such data. Furthermore, many fragment ions are simply ignored during data analysis, in large part because automated search engines do not ‘look’ for all potential fragmentation products, and also because we simply still do not sufficiently understand the mechanisms of gas-phase peptide fragmentation to fully interpret the spectra (most likely a combination of the two). The end result is that even though proteome coverage is increasing in large-scale analyses, we are still a long way from the ideal of ‘complete’ proteome analysis.
This webinar showcases the Growth Direct System; an RMM (Rapid Microbial Method) that improves on traditional membrane filtration, delivering increased accuracy, a faster time to result, enhanced data integrity compliance, and more control over the manufacturing process.
Key learning points:
Understand the benefits of full workflow microbiology quality control testing automation in radiopharmaceutical production
Learn about ITM’s implementation journey and considerations when evaluating the technology
Find out how the advanced optics and microcolony detection capabilities of Growth Direct® technology impact time to result (TTR).
Don’t miss your chance to learn from experts in the industry –Register for FREE
Can’t attend live? No worries – register to receive the recording post-event.
Mass spectrometry (MS) has become a pivotal analytical technique for proteomic studies. Individual proteins within complex mixtures can be identified through matching of experimentally acquired peptide fragmentation spectra to known protein sequences based on the well-understood principles of peptide ion fragmentation2. In general, the presence of an expressed gene product is ascertained by MS/MS analysis of peptides rather than proteins, because the complications that arise from interpreting tandem mass spectra of highly charged proteins and/or multiple covalent protein modifications are minimised. Such post-translational modifications cause a combinatorial ‘explosion’ generating a myriad of isoforms that complicate data analysis and reduce signal intensity for the individual protein species3. ‘Identification’ of proteins present in a complex mixture is thus inferred following interrogation of peptide MS/MS data using dedicated search engines including MASCOT and SEQUEST4-7.
Forgotten protein fragments
As indicated previously, the vast majority of protein ‘identification’ is inferred by MS/MS analysis of proteolytically derived peptides. However, in the type of data-dependent analysis (DDA) typically performed, only the n most intense peptide ions (where n is typically 3-8) are isolated and subjected to MS/MS and structural characterisation. The result is that a number of the proteolytic fragments derived from a single protein (those that have reduced ionisation efficiency and are thus less likely to be observed in a reverse-phase liquid chromatography (LC)-ESI experiment8) are ‘invisible’ in the MS/MS experiment and therefore cannot be used to help confirm presence of an expressed gene product. In contrast, data-independent analysis (DIA) such as that performed in an MSE workflow9, where all peptide ions that are generated at a given time following LC elution are subjected to collision-induced dissociation (CID), potentially makes use of all peptide ions for confirming protein expression. This type of experiment can thus result in significant increases in the number of confidently identified peptides (and by inference, confidently identified proteins) when compared to a more standard DDA analysis10.
‘Forgotten’ spectra
A major surprise to many scientists entering the proteomics field is the amount of data acquired that is ultimately ‘wasted’. Of the thousands of tandem mass spectra that may be acquired in a typical DDA experiment, only a proportion (~
‘Forgotten’ fragment ions
An additional problem arises with the way in which most search engines function when analysing tandem MS data. Often only a small proportion of the fragment ions present within a given spectrum are utilised. Important information that could enhance data analysis, such as fragment ion intensities, internal ions and neutral loss from precursor ions, are often neglected13. The presence of non-sequence, or ‘scrambled’ N-terminally derived b-ions (albeit at a low level), which have also been observed in spectra following CID are also not taken into consideration14-16. Further complications may arise during data analysis as the majority of search engines are even less well adapted to the analysis of spectra arising from alternate fragmentation methods such as Electron Capture Dissociation (ECD) or Electron Transfer Dissociation (ETD). A greater understanding of the underlying mechanistic principles of peptide dissociation and perhaps more crucially, the incorporation of these principles into the search engines employed in automated data analysis is therefore a challenge yet to be addressed.
Sequence scrambled fragments
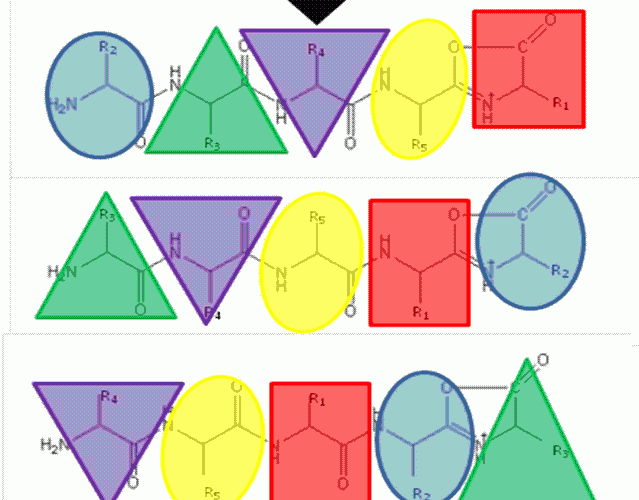
The ‘Mobile Proton’ model from the mid- 1990’s17,18 describes the mechanistic basis for peptide fragmentation by CID, which results in the production of b- and y-ions from the amino- and carboxy- termini of the peptide respectively. Subsequent neutral loss of CO from b-ions can also result in the formation of a-ions. It is these ions (together with neutral loss of water or ammonia from amino acid side-chains) that are typically used for sequence identification19,20. However, CID spectra often contain ions that cannot be assigned by solely considering these classical fragmentation pathways. The formation of a macrocycle intermediate which can occur during b-ion formation may result in sequence ‘scrambling’ if ring opening occurs at an ‘internal’ peptide bond14. Under these circumstances, N-terminal amino acid(s) are displaced towards the C-terminus, exposing previously internal amino acids at the terminus (Figure 1). These rearranged b-ions can then undergo further fragmentation according to con – ventional CID pathways, resulting in a fragment ion series derived from the non-native peptide sequence, which significantly complicates spectral interpretation.
FIGURE 1The b5 ion is generated by CID which terminates at the C-terminus in an oxazolone ring structure. N-terminal nucleophilic attack of the oxazolone ring results in formation of a macrocyclic intermediate that undergoes ring opening at various positions to generate sequence scrambled b5 ions.
Recently, research has been undertaken to identify factors that either promote or inhibit bion scrambling, with amino acid composition/ residue position15,21-24, and sequence length14,25,26 having all been considered. However, to fully understand the underlying mechanisms of peptide scrambling, several questions remain unanswered. Current research has primarily focused on synthesised ‘model’ peptides to address specific issues. To date, the most notable findings include the lack of observable effect on scrambling (positive or negative) as a result of the presence or position of acidic residues21 and an inhibition of macrocycle formation (and thus the potential for sequence rearrangement) intrinsic to peptides containing an internal arginine residue22. Additionally it has been demonstrated that N-terminal acetylation of a peptide compromises sequence scrambling16. It is thought that the presence of an acetyl group prevents nucleophilic attack of the oxazolone ring and thus macrocycle formation/ring opening is avoided. Figure 2 shows the effect of N-acetylation on the CID tandem mass spectrum of human β-casomorphin (YPFVEPI). The modification simplifies the spectrum (Figure 2A) as it lacks the sequence scrambled product ions generated in its absence (Figure 2B).
FIGURE 2 Collision-Induced Dissociation of (A) N-acetylated β-casomorphin (Ac-YPFVEPI) and (B) CID of β-casomorphin (YPFVEPI)
Internal fragment ions
Collisional activation of protonated peptides generates product ions resulting from both simple bond cleavages27 and complex, multistage rearrangements14. In addition, depending upon the instrument platform and the energy of the collisional activation employed, fragments arising from multiple bond cleavages can be observed with abundant signal intensities in MS/MS spectra. Such fragments are termed ‘internal ions’ and result from charge retention on an excised section of the peptide backbone. Formation of internal fragment ions arises from a combination of either b-/y-type cleavage (amino-acylium ion) or a-/y-type cleavage (amino-immonium ion). It is important to acknowledge the potential formation of such fragments to avoid the mis-assignment of internal ions as those arising from a single bond cleavage. Work by Gaskell and co-workers28 demonstrated a greater relative abundance of these internal fragment ions in low energy fragmentation regimes. The increased observation of these internal fragment ions in quadrupole collision cells can largely be attributed to the extended timescale of the activation/decomposition process and the greater potential for multiple ion-neutral collisions, which increases the prevalence of multiple bond cleavages. Other internal ions, commonly termed immonium ions, are also frequently observed; these appear in the low mass region of MS/MS spectra, usually representing an amino acid located internally within the peptide backbone. Interestingly, although observation of these immonium ions is commonly used in manual interpretation to infer the presence of specific amino acids in the peptide sequence, this information is not exploited by search algorithms.
Hydrogen atom abstraction products in Electron Transfer Dissociation (ETD)
Electron Transfer Dissociation (ETD; Figure 3) is especially useful for fragmentation of large, highly charged peptides or when preservation of potentially labile side-chains or posttranslational modifications is required29-32. For example, labile phosphate groups can be lost from amino acid side-chains during CID, making pinpointing the modification site(s) challenging33. Using ETD, a molecular anion is used to transfer an electron to a peptide cation, thereby inducing fragmentation at the N-Cα bond34-35. ETD is typically utilised on ion trap mass spectrometers but has also been successfully used on a range of instrumental platforms36-38. Cleavage of peptide chains by ETD primarily proceeds in a non-specific manner and the size of residue and the amino acid sequence do not in general affect the process. An exception to this rule is that bond cleavage is not observed N-terminal to the imino acid proline, which lacks a hydrogen on the amide group and cannot consequently participate in ETD fragmentation.
FIGURE 3Mechanism of fragmentation during Electron Transfer Dissociation
During ETD, fragmentation generating c+ and z+. ions is typical. However both c+. and z+ ions can also be observed, although their contribution is thought to be of much less significance, particularly under ‘standard’ ETD conditions. Interestingly, Coon et al. have reported increased propensity for the formation of odd electron c+. and even electron z+ species when employing Electron Transfer Dissociation in combination with non-dissociative Collision Activated Dissociation (ETcaD)39. This technique, known as ‘supplemental activation’, involves subjecting a non-dissociated electron transfer product [M+nH](n-1)+. to low-energy collisional activation that lies below the threshold required to produce fragmentation by CID. The mechanism of hydrogen atom abstraction which forms these c+. and z+ ions is believed to be similar to that previously described for Electron Capture Dissociation (ECD)40-42. Newly formed c/z. ions associate through non-covalent interactions (e.g. hydrogen bonds) and the extended lifetime of such complexes facilitates inter-molecular hydrogen abstraction to generate c./z ions. These newly formed c. and z ions have a mass difference of plus and minus 1 Da as compared to the original c anc z. ions, respectively. Supplemental activation helps to break this dissociation complex, resulting in the highly efficient production of electron transfer products. Although search algorithms do consider hydrogen abstraction products, little is known about the underlying mechanism of the transfer process. While it is trivial to predict the observed resultant mass shift, further studies are required to understand how the peptide sequence and the timescale and energetics of supplemental activation could influence the extent of hydrogen transfer. Enhanced knowledge of these factors would enable prediction of relative fragment ion intensities, specifically the apparent change in isotope ratios that are observed as a result of isotopic envelope overlap of the original c anc z. ions their hydrogen abstracted products. Ultimately, this would enable more informed database searches to be performed with ETD generated data, yielding better results than is presently possible.
Summary
Mass spectrometry has a central, expanding role to play in proteomic analysis and protein biomarker discovery, and it is evident that an ever-increasing quantity of high-quality information will continue to be generated in the future. However, addressing fundamental issues surrounding various ‘missing fragments’, be they protein fragments ‘lost’ to analysis, or fragment ion information ‘lost’ during data interpretation is a major challenge in the field. Indeed, by improving basic knowledge surrounding the mechanisms of peptide ionisation and fragmentation, and ensuring it’s implementing into analytical pipelines, there should be vast potential for significantly improving the information derived from MS-based proteomics analysis in the future. Remarkably, the MS community has been studying peptide fragmentation in the gas-phase for nearly 50 years; however, our ability to fully understand and interpret a peptide ion derived tandem mass spectrum remains incomplete. Hopefully, it will not take another half century to fully tap the enormous wealth of information being generated worldwide.
Acknowledgements
RC thanks the EPSRC for a CASE funded PhD Studentship. CEE acknowledges the Royal Society for a Dorothy Hodgkin Research Fellowship and the BBSRC for funding (BB/H007113/1).
References
1. Michalski, A., J. Cox, and M. Mann, More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J Proteome Res, 2011. 10(4): p. 1785-93
2. Mallick, P., et al., Precursor-ion mass re-estimation improves peptide identification on hybrid instruments. Journal of Proteome Research, 2008. 7(9): p. 4031-4039
3. Schweppe, R.E., et al., The characterization of protein post-translational modifications by mass spectro – metry. Accounts of Chemical Research, 2003. 36(6): p. 453-461
4. Krijgsveld, J., et al., In-gel isoelectric focusing of peptides as a tool for improved protein identification. Journal of Proteome Research, 2006. 5(7): p. 1721- 1730
5. Pappin, D.J.C., et al., Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis, 1999. 20(18): p. 3551-3567
6. Eng, J.K., A.L. Mccormack, and J.R. Yates, An Approach to Correlate Tandem Mass-Spectral Data of Peptides with Amino-Acid-Sequences in a Protein Database. Journal of the American Society for Mass Spectrometry, 1994. 5(11): p. 976-989
7. Wedge, D.C., et al., FDRAnalysis: a tool for the integrated analysis of tandem mass spectrometry identification results from multiple search engines. J Proteome Res, 2011. 10(4): p. 2088-94
8. Eyers, C.E., et al., CONSeQuence: prediction of reference peptides for absolute quantitative proteomics using consensus machine learning approaches. Mol Cell Proteomics, 2011
9. Silva, J.C., et al., Absolute quantification of proteins by LCMSE – A virtue of parallel MS acquisition. Molecular & Cellular Proteomics, 2006. 5(1): p. 144-156
10. Blackburn, K., et al., Improving protein and proteome coverage through data-independent multiplexed peptide fragmentation. J Proteome Res, 2010. 9(7): p. 3621-37
11. Junqueira, M., et al., Separating the wheat from the chaff: unbiased filtering of background tandem mass spectra improves protein identification. J Proteome Res, 2008. 7(8): p. 3382-95
12. Lee, K.A., et al., 24-Hour Lock Mass Protection. Journal of Proteome Research, 2011. 10(2): p. 880-885
13. Baldwin, M.A., Protein identification by mass spectrometry – Issues to be considered. Molecular & Cellular Proteomics, 2004. 3(1): p. 1-9
14. Vazquez, J., et al., Peptide rearrangement during quadrupole ion trap fragmentation: Added complexity to MS/MS spectra. Analytical Chemistry, 2003. 75(6): p. 1524-1535
15. Gaskell, S.J., et al., Evidence for structural variants of a- and b-type peptide fragment ions using combined ion Mobility/Mass spectrometry. Journal of the American Society for Mass Spectrometry, 2008. 19(4): p. 609-613
16. Harrison, A.G., Peptide Sequence Scrambling Through Cyclization of b(5) Ions. Journal of the American Society for Mass Spectrometry, 2008. 19(12): p. 1776-1780
17. Dongre, A.R., et al., Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: Evidence for the mobile proton model. Journal of the American Chemical Society, 1996. 118(35): p. 8365-8374
18. Wysocki, V.H., et al., Special feature: Commentary – Mobile and localized protons: a framework for understanding peptide dissociation. Journal of Mass Spectrometry, 2000. 35(12): p. 1399-1406
19. Mueller, D.R., M. Eckersley, and W.J. Richter, Hydrogen Transfer-Reactions in the Formation of Y+2 Sequence Ions from Protonated Peptides. Organic Mass Spectrometry, 1988. 23(3): p. 217-222
20. Cordero, M.M., J.J. Houser, and C. Wesdemiotis, The Neutral Products Formed during Backbone Fragmentations of Protonated Peptides in Tandem Mass-Spectrometry. Analytical Chemistry, 1993. 65(11): p. 1594-1601
21. Yalcin, T. and A.E. Atik, A Systematic Study of Acidic Peptides for b-Type Sequence Scrambling. Journal of the American Society for Mass Spectrometry, 2011. 22(1): p. 38-48
22. Van Stipdonk, M., S. Molesworth, and S. Osburn, Influence of Amino Acid Side Chains on Apparent Selective Opening of Cyclic b(5) Ions. Journal of the American Society for Mass Spectrometry, 2010. 21(6): p. 1028-1036
23. Van Stipdonk, M., et al., Sequence-Scrambling Fragmentation Pathways of Protonated Peptides. Journal of the American Chemical Society, 2008. 130(52): p. 17774-17789
24. Harrison, A.G., et al., Effect of the His Residue on the Cyclization of b Ions. Journal of the American Society for Mass Spectrometry, 2010. 21(8): p. 1352-1363
25. Gaskell, S.J., et al., Studies of Peptide a- and b-Type Fragment Ions Using Stable Isotope Labeling and Integrated Ion Mobility/Tandem Mass Spectrometry. Journal of the American Society for Mass Spectrometry, 2008. 19(12): p. 1781-1787
26. Polfer, N.C., et al., Effect of Peptide Fragment Size on the Propensity of Cyclization in Collision-Induced Dissociation: Oligoglycine b(2)-b(8). Journal of the American Chemical Society, 2009. 131(51): p. 18272-18282
27. Roepstorff, P. and J. Fohlman, Proposal for a Common Nomenclature for Sequence Ions in Mass-Spectra of Peptides. Biomedical Mass Spectrometry, 1984. 11(11): p. 601-601
28. Ballard, K.D. and S.J. Gaskell, Sequential Mass- Spectrometry Applied to the Study of the Formation of Internal Fragment Ions of Protonated Peptides. International Journal of Mass Spectrometry and Ion Processes, 1991. 111: p. 173-189
29. Hunt, D.F., et al., Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America, 2004. 101(26): p. 9528-9533
30. Hunt, D.F., et al., The utility of ETD mass spectrometry in proteomic analysis. Biochimica Et Biophysica Acta- Proteins and Proteomics, 2006. 1764(12): p. 1811-1822
31. Hunt, D.F., et al., Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America, 2007. 104(7): p. 2193-2198
32. Chalkley, R., O-GlcNAcylation: The Post-Translational Modification that Best Highlights the Value of ETD. Molecular & Cellular Proteomics, 2009: p. S13-S13
33. Sickmann, A., J. Wiesner, and T. Premsler, Application of electron transfer dissociation (ETD) for the analysis of posttranslational modifications. Proteomics, 2008. 8(21): p. 4466-4483
34. Shabanowitz, J., et al., Analysis of intact proteins on a chromatographic time scale by electron transfer dissociation tandem mass spectrometry. International Journal of Mass Spectrometry, 2007. 259(1-3): p. 197-203
35. Turecek, F., X.H. Chen, and C.T. Hao, Where does the electron go? Electron distribution and reactivity of peptide cation radicals formed by electron transfer in the gas phase. Journal of the American Chemical Society, 2008. 130(27): p. 8818-8833
36. Mann, M., et al., A Dual Pressure Linear Ion Trap Orbitrap Instrument with Very High Sequencing Speed. Molecular & Cellular Proteomics, 2009. 8(12): p. 2759-2769
37. Williams, J.P., et al., Identifying drug metallation sites on peptides using electron transfer dissociation (ETD), collision induced dissociation (CID) and ion mobility-mass spectrometry (IM-MS). Chemical Communications, 2010. 46(30): p. 5458-5460
38. Hartmer, R. and M. Lubeck, New approach for characterization of post translational modified peptides using ion trap MS with combined ETD/CID fragmentation. Lc Gc Europe, 2005: p. 11-13.
39. Coon, J.J., et al., Supplemental activation method for high-efficiency electron-transfer dissociation of doubly protonated peptide precursors. Analytical Chemistry, 2007. 79(2): p. 477-485
40. Turecek, F. and E.A. Syrstad, Mechanism and energetics of intramolecular hydrogen transfer in amide and peptide radicals and cation-radicals. Journal of the American Chemical Society, 2003. 125(11): p. 3353-3369
41. Fung, Y.M.E. and T.W.D. Chan, Experimental and theoretical investigations of the loss of amino acid side chains in electron capture dissociation of model peptides. Journal of the American Society for Mass Spectrometry, 2005. 16(9): p. 1523-1535
42. McLafferty, F.W., et al., Electron capture dissociation of gaseous multiply charged ions by Fourier-transform ion cyclotron resonance. Journal of the American Society for Mass Spectrometry, 2001. 12(3): p. 245-249
About the Authors
Dr. Claire Eyers is a Royal Society Dorothy Hodgkin Research Fellow and Acting Director of the Michael Barber Centre for Mass Spectrometry at the University of Manchester. Having received her PhD from the University of Dundee in 2002 under the guidance of Professor Sir Philip Cohen, she moved to the University of Colorado at Boulder to further her interests in mass spectrometric analysis of protein phosphorylation. Having initially relocated to the University of Manchester to pursue postdoctoral research with Professor Simon Gaskell, since 2007 she has managed her own research group focusing on the application of biophysical and biochemical techniques to study cellular protein phosphorylation at a global level.
Ross Chawner is a postgraduate student at the University of Manchester where he is undertaking a PhD sponsored by EPSRC and Waters. He received his BSc in Applied Chemistry from Manchester Metropolitan University in 2008 while working for AstraZeneca, where he completed an apprenticeship in the Separation Science Laboratory before moving to the Physical Chemistry Department performing Lipophilicity and Protein Binding Measurements. His current research focuses on elucidation of peptide fragmentation by Ion Mobility Mass Spectrometry.
This website uses cookies to enable, optimise and analyse site operations, as well as to provide personalised content and allow you to connect to social media. By clicking "I agree" you consent to the use of cookies for non-essential functions and the related processing of personal data. You can adjust your cookie and associated data processing preferences at any time via our "Cookie Settings". Please view our Cookie Policy to learn more about the use of cookies on our website.
This website uses cookies to improve your experience while you navigate through the website. Out of these cookies, the cookies that are categorised as ”Necessary” are stored on your browser as they are as essential for the working of basic functionalities of the website. For our other types of cookies “Advertising & Targeting”, “Analytics” and “Performance”, these help us analyse and understand how you use this website. These cookies will be stored in your browser only with your consent. You also have the option to opt-out of these different types of cookies. But opting out of some of these cookies may have an effect on your browsing experience. You can adjust the available sliders to ‘Enabled’ or ‘Disabled’, then click ‘Save and Accept’. View our Cookie Policy page.
Necessary cookies are absolutely essential for the website to function properly. This category only includes cookies that ensures basic functionalities and security features of the website. These cookies do not store any personal information.
Cookie
Description
cookielawinfo-checkbox-advertising-targeting
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertising & Targeting".
cookielawinfo-checkbox-analytics
This cookie is set by GDPR Cookie Consent WordPress Plugin. The cookie is used to remember the user consent for the cookies under the category "Analytics".
cookielawinfo-checkbox-necessary
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
This cookie is set by GDPR Cookie Consent WordPress Plugin. The cookie is used to remember the user consent for the cookies under the category "Performance".
PHPSESSID
This cookie is native to PHP applications. The cookie is used to store and identify a users' unique session ID for the purpose of managing user session on the website. The cookie is a session cookies and is deleted when all the browser windows are closed.
viewed_cookie_policy
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
zmember_logged
This session cookie is served by our membership/subscription system and controls whether you are able to see content which is only available to logged in users.
Performance cookies are includes cookies that deliver enhanced functionalities of the website, such as caching. These cookies do not store any personal information.
Cookie
Description
cf_ob_info
This cookie is set by Cloudflare content delivery network and, in conjunction with the cookie 'cf_use_ob', is used to determine whether it should continue serving “Always Online” until the cookie expires.
cf_use_ob
This cookie is set by Cloudflare content delivery network and is used to determine whether it should continue serving “Always Online” until the cookie expires.
free_subscription_only
This session cookie is served by our membership/subscription system and controls which types of content you are able to access.
ls_smartpush
This cookie is set by Litespeed Server and allows the server to store settings to help improve performance of the site.
one_signal_sdk_db
This cookie is set by OneSignal push notifications and is used for storing user preferences in connection with their notification permission status.
YSC
This cookie is set by Youtube and is used to track the views of embedded videos.
Analytics cookies collect information about your use of the content, and in combination with previously collected information, are used to measure, understand, and report on your usage of this website.
Cookie
Description
bcookie
This cookie is set by LinkedIn. The purpose of the cookie is to enable LinkedIn functionalities on the page.
GPS
This cookie is set by YouTube and registers a unique ID for tracking users based on their geographical location
lang
This cookie is set by LinkedIn and is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
lidc
This cookie is set by LinkedIn and used for routing.
lissc
This cookie is set by LinkedIn share Buttons and ad tags.
vuid
We embed videos from our official Vimeo channel. When you press play, Vimeo will drop third party cookies to enable the video to play and to see how long a viewer has watched the video. This cookie does not track individuals.
wow.anonymousId
This cookie is set by Spotler and tracks an anonymous visitor ID.
wow.schedule
This cookie is set by Spotler and enables it to track the Load Balance Session Queue.
wow.session
This cookie is set by Spotler to track the Internet Information Services (IIS) session state.
wow.utmvalues
This cookie is set by Spotler and stores the UTM values for the session. UTM values are specific text strings that are appended to URLs that allow Communigator to track the URLs and the UTM values when they get clicked on.
_ga
This cookie is set by Google Analytics and is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. It stores information anonymously and assign a randomly generated number to identify unique visitors.
_gat
This cookies is set by Google Universal Analytics to throttle the request rate to limit the collection of data on high traffic sites.
_gid
This cookie is set by Google Analytics and is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visited in an anonymous form.
Advertising and targeting cookies help us provide our visitors with relevant ads and marketing campaigns.
Cookie
Description
advanced_ads_browser_width
This cookie is set by Advanced Ads and measures the browser width.
advanced_ads_page_impressions
This cookie is set by Advanced Ads and measures the number of previous page impressions.
advanced_ads_pro_server_info
This cookie is set by Advanced Ads and sets geo-location, user role and user capabilities. It is used by cache busting in Advanced Ads Pro when the appropriate visitor conditions are used.
advanced_ads_pro_visitor_referrer
This cookie is set by Advanced Ads and sets the referrer URL.
bscookie
This cookie is a browser ID cookie set by LinkedIn share Buttons and ad tags.
IDE
This cookie is set by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
li_sugr
This cookie is set by LinkedIn and is used for tracking.
UserMatchHistory
This cookie is set by Linkedin and is used to track visitors on multiple websites, in order to present relevant advertisement based on the visitor's preferences.
VISITOR_INFO1_LIVE
This cookie is set by YouTube. Used to track the information of the embedded YouTube videos on a website.