Concerns around Annex 1
Posted: 21 May 2019 | Dr Tony Cundell - Microbiological Consulting LLC | No comments yet
This article expresses the opinions of a pharmaceutical microbiologist on the proposed revisions to the EU Good Manufacturing Practice Annex 1 in terms of current industry practice and future innovation in sterile product manufacturing.

As a microbiological consultant working in the pharmaceutical industry, my reaction to the revisions to EU Guidelines to Good Manufacturing Practice – Medicinal Products for Human and Veterinary Use Annex 1. Manufacture of Sterile Medicinal Products was one of disquiet. Although we expect regulatory science to evolve, there is always a tension between industry best practice and the positions taken by regulators. Much of the Annex’s content is, in my opinion, prescriptive, unsupported by technical literature, will not promote a broader understanding of sterile drug manufacturing and may discourage risk analysis.
My major concerns fall into the following four areas:
- Requirements in Annex 1 becoming the de facto rules that govern sterile product manufacturing globally when the revised Annex contains requirements that do not represent industry best practice and conflict with other regulatory guidance
- Setting microbial requirements for air, surfaces and personnel by air cleanliness classifications of aseptic process environments when they cannot be engineering standards and when these environments vary greatly in design, operation and potential risk of product contamination
- Failure to recognise the poor analytical capabilities of current microbial monitoring methods
- Continuing to support the equivalency requirement for new microbiological technologies that are not dependent of microbial growth and the colony-forming unit.
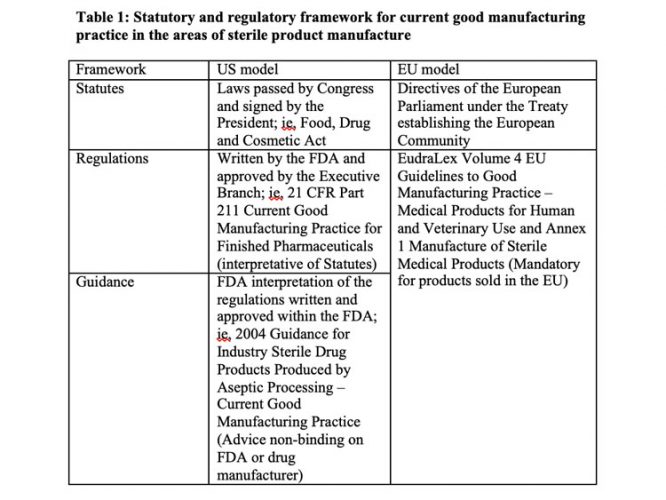
Other US viewpoints on the Annex 1 revision from experts in the area of aseptic processing and engineering were published last year.1 Table 1 describes the regulatory framework for GMPs in the US and EU.
The EU GMPs were first published in 1989, reconstructed in October 2005 and updated in December 2010. I believe that Annex 1 dates back to September 2003 and has been revised to align the cleanroom classification table and provide guidance on media simulations, bioburden monitoring and the capping of vials. The current extensive revision published in December 2017 and subject to considerable comment, will be official when finalised. The guidelines are unlikely to be revised again before the passage of a decade. With a “how to” and not a “what to do” approach, the pharmaceutical industry is bound to these mandatory requirements, which will have a dead hand on any advances in manufacturing and monitoring technologies.
The guide is presented in three parts and supplemented by a series of annexes, of which Annex 1. Manufacture of Sterile Medicinal Products is the topic of this article.
The EU Guideline is divided into:
- Part I: covering GMP principles for the manufacture of medicinal products
- Part II: covering GMP for active substances used as starting materials
- Part III: containing GMP-related documents, which clarify regulatory expectations.
These parts of the EU GMPs are high-level requirements comparable to those found in 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceuticals.
Differences in sterile product current good manufacturing practice
The 2004 FDA Guidance for Industry Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice is not enforceable but recommends how a sterile drug manufacturer can comply with the cited sections of the GMP regulations and allows other approaches, as justified. In contrast, Annex 1 regulations must be complied with if the manufacturer wants to market their products in the EU. The revision contains many requirements that are not industry practice and are not scientifically justified. I recommend reviewing the recently published 2017 PDA Aseptic Processing Survey to benchmark industry practice.2
The revision was undertaken by a joint EMA and PIC/S working group that represents a broad base of multi-national pharmaceutical manufacturing experience. The Annex 1 revision may make mutual recognition of GMP compliance inspections between the EMA and the FDA more difficult as there are unresolved differences in how to achieve GMP compliance between the two regulatory agencies. Both agencies will retain and exercise the option to conduct separate investigations for cause and pre-approval inspections, where the inspectional differences will be most clearly manifested with one agency taking regulatory action while the other may see no compliance issues. If manufacturing sites outside the EU cannot comply with these changes, this will increase drug shortages in the EU.
Setting microbial requirements by air cleanliness classifications
Setting air cleanliness standards for the engineering design and operation of an aseptic process environment based on particulate concentrations, air velocities, air changes, space pressurisation, temperature and relative humidity makes sense; whereas setting microbial monitoring standards does not, when often they are area use and people related. Aseptic processing environments vary widely in terms of their risk to microbial product contamination from laminar flow hoods to isolator systems. The ability to exclude people from aseptic processing has a hierarchy of risk – laminar flow hood > biological safety cabinets > restricted access barrier systems > open isolator systems > closed isolator systems with gloves > gloveless isolator systems – that is not fully recognised in Annex 1.
Analytical capabilities of current microbial monitoring methods
It is my experience that colleagues in the pharmaceutical industry who are not microbiologists expect too much of microbial monitoring methods, giving more creditability to the plate counts than is justified. These issues are more fundamental than the incubation conditions of environmental monitoring plates and media fills highlighted by the PDA review of the EU GMP Annex 1. Our methods are unreliable for the following reasons:
- Microorganisms are never uniformly distributed in the environment so reliable sampling is always an issue, especially at microbial densities
- Most microorganisms will not grow on standard microbiological growth media. The term “the great plate count anomaly” was coined by Staley and Konopka to describe the difference in orders of magnitude between the numbers of cells from natural environments that form colonies on agar media and the numbers countable by microscopic examination3
- As microorganisms in manufacturing facilities are frequently associated with shed skin cells, water droplets or dust particles, a colony‑forming unit is most likely not derived from a single microbial cell
- Microbial levels are set below the limit of quantification where the accuracy and precision of the microbial counts are poor.
Equivalency requirement of new microbiological technologies
The USP general informational chapter <1223> Validation of Alternative Microbiological Methods contains a discussion of the limitations of the colony-forming unit and why microbial detection and enumeration methods that employ different signals to the colony-forming unit may not be equivalent to traditional methods. The signals may include laser-induced fluorescent particles, ATP bioluminescence, enzymic activity, headspace analysis and genomic weights. This was addressed in <1223> by broadening the validation options used to qualify an alternative microbiological method.
Four options are available to establish the equivalence of a candidate alternative analytical method: acceptable procedures (ie, merely meeting a minimum performance or acceptance requirement without a need to demonstrate equivalence to the compendial method); performance equivalence to the compendial method; results equivalence to the compendial method; and decision equivalence to the compendial method.
For example, light-induced fluorescent viable particle monitoring of air and pharmaceutical-grade water can provide continuous in-process monitoring data that is not equivalent to daily or weekly monitoring using colony-forming units obtained from plate counts but may be used to make superior decisions related to a loss of microbial control or adverse trends during sterile product manufacturing. As results are not equivalent, different alert/action levels and trending rules would need to be established, resulting in an improved level of environmental and operational control.4,5,6 The positions taken in the revision to Annex 1 prevent the industry from taking advantage of this new technology.
Furthermore, real-time analysis of the in-process microbiological air quality is consistent with the principles of ICH Q8 Quality Risk Management (QRM), ICH Q9 Quality by Design (QbD) and Process Analytical Technology (PAT).
Conclusions
The working group needs to re-visit the proposed version and make additional changes.
Biography
Dr Tony Cundell has a PhD in Microbiology from the Lincoln University, New Zealand. He consults in the areas of microbial risk assessment, regulatory affairs and microbiological testing. Prior to November 2013 he worked for Merck Research Laboratories in Summit, New Jersey as the Senior Principal Scientist in early phase drug development. Earlier in his career, Tony worked at a director level in Quality Control and Product Development organisations at the New York Blood Center, Lederle Laboratories, Wyeth Pharmaceuticals and Schering-Plough. He is a member of the 2015‑2020 U.S.P. Microbiology Committee of Experts where he takes a leadership role in the area of modern microbiology methods.
References
1. JP. 2018 Annex 1 Misses the Mark pharmtech.com/annex-1-misses-mark-expanded-version March 14 2018
2. PDA Aseptic Processing Survey December 2017, pp169
3. Staley JT, Konopka A. 1985 Measurement of in situ activities of non-photosynthetic microorganisms in aquatic and terrestrial habitats. Ann. Rev Microbiol. 39:321-46.
4. Miller MJ, Walsh MR, Shrake JL, Dukes RE, Hill DB. 2009 Evaluation of the Biovigilant IMD-A, a Novel Spectroscopy Technology for the Continuous and Real-time Monitoring of Viable and Non-viable Particles. Part 2: Case Studies in Environmental Monitoring during Aseptic Filling, Intervention Assessments, and Glove Integrity Testing in Manufacturing Isolators PDA J. Pharm. Sci. & Technol. 63(3): 259-283.
5. Cundell AM. (Workgroup Member) Novel Concept for Online Water Bioburden Analysis: Key Considerations, Applications, and Business Benefits for Microbiological Risk Reduction. Amer. Pharm. Review. Aug.-Sept. 2013 issue, Specification Edition pp 4-7 2013.
6. Hutchins PM. 2016 Real-Time Viable Particle Monitoring: Principles and Benefits for In-Process Measurements. Am. Pharm. Rev. Nov.-Dec. 2016 issue. www.americanpharmaceuticalreview.com
Issue
Related topics
Aseptic Processing, Good Manufacturing Practice (GMP), Microbial Detection, Microbiology, Regulation & Legislation
Related organisations
European Medicines Agency (EMA), US Food and Drug Administration (FDA)