Does the Falsified Medicines Directive lead to a European pack coding guideline?
Posted: 22 April 2017 | Johan Verhaeghe (Medicines for Europe) | No comments yet
The Directive on Falsified Medicines (FMD, or Directive 2011/62/EU amending Directive 2001/83/EC) and its supplementing Delegated Regulation (DR 2016/161) aim to improve patient safety by preventing falsified medicines from entering the legal supply chain…

One measure intended to achieve this goal is mandating marketing authorisation holders and manufacturers of medicinal products (for human use) to apply safety features to the outer packaging of medicinal products. These safety features are an anti-tampering device and a unique identifier (UI) that allows products’ authenticity to be verified at the point of dispense.
To enable compliance with the FMD requirements (focusing on the verification of the UI), the following industry bodies have established the European Medicines Verification Organisation (EMVO): the European Federation of Pharmaceutical Industries and Associations (EFPIA – representing the research-based pharmaceutical industry); Medicines for Europe (representing the generic, biosimilar and value-added medicines industries); the European Association of Euro-Pharmaceutical Companies (EAEPC – representing the parallel distribution industry); the Pharmaceutical Group of the European Union (PGEU – representing community pharmacists), and the umbrella organisation of pharmaceutical full-line wholesalers in Europe (GIRP).
The EMVO has taken responsibility for advancing the formation of the European Medicines Verifications System (EMVS) in order to achieve the implementation of a functioning, secure, interoperable and cost-effective system across Europe by February 2019 that will, as mentioned, allow the point-of-dispense verification of the UI in accordance with the FMD and its DR (see Figure 1).
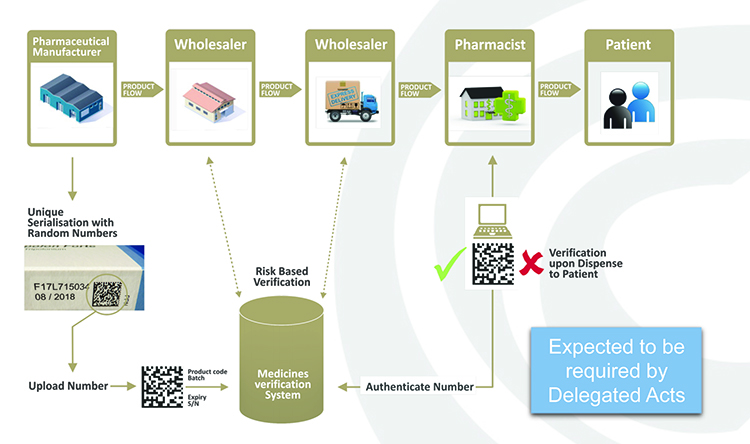
Figure 1: Point-of-dispense verification.
In practice, manufacturers will be required to print a data matrix code, which incorporates a UI, and apply an anti-tampering device on the outer packaging of (mainly) prescription medicines for each individual sales package. At the point of dispense the medicine will be scanned and verified for authenticity against a national (or supranational) repository (see Figure 2). If the UI on the pack matches the information in the repository, the UI is ‘decommissioned’ and the pack can be supplied to the patient. If there is a warning related to this UI, the system will highlight this as an exceptional event and the pack will not be supplied to the patient. An investigation needs to determine whether the pack has been falsified or not.
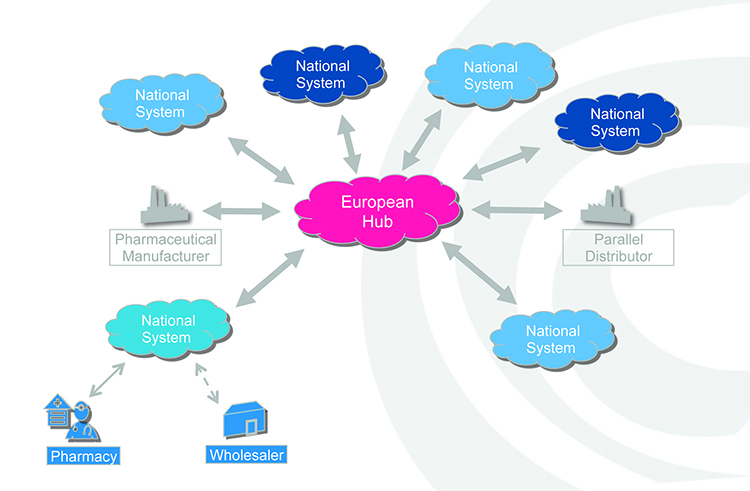
Figure 2: Authenticity checking against a national or supranational hub.
Coding and printing
The use of the UI has various consequences. Firstly, the design of the pack might change. This would not automatically be the case but the dimensions of the UI differ from a standard or linear barcode and thus a change is most likely and expected. Secondly, the UI has mandatory lines of code in human-readable format that need to be printed adjacent to the 2D matrix. In practice this results in at least two more lines to be printed: the serial number and product code (the expiry date and batch number are already printed). Again, exceptions are foreseen but the expectation is that in practice this will change the design of the majority of packages. Furthermore, national competent authorities may require the printing of a national health reimbursement number (NHRN).
Regulatory pathway
This overall process where a barcode is being replaced by a 2D data matrix and its at least four human readable elements will change the layout of a medicinal pack. The manufacturers could consequently be required to apply for a variation or notification procedure.
At times this can be done for the European area via a centralised procedure, but most products are registered at a national level. A quick look at data from the European Medicines Agency tells us there are approximately 400,000 marketing authorisations (MA) which are subject to the FMD and thus require a change to the layout of the outer package.
A variation or notification procedure requires not only an investment from the industry side (rethink the artwork, adapt the printing, costs for submission of the files per MA, etc), the authorities also face a huge workload as all these applications need to be processed. Not only will the procedure per application take some time, the workload of processing many applications in a short time period could (and unfortunately most likely will) lead to bottlenecks.
Multi-market packs
Multi-market packs are those that are specifically made to be sold in more than one market. Consequently, the overall design is similar for more than one country. If the layout of such a pack changes due to a national specific requirement, this would also need to be acceptable for every other country where this multi-market pack would be sold. For example, a pack that can be sold in France and Belgium for which the packaging design must be changed due to a new requirement for the French market can no longer be sold in Belgium unless Belgium also approves the proposed change.
If a country requires a human-readable NHRN on pack, this pack becomes country specific virtually by default. As in the example above, this would mean that where multi-market packs exist and one of the countries required a national code in a human readable format, other potential markets would need to allow this code to be printed.
For example, if France required an on-pack NHRN for a product originally also sold in Belgium, then the French NHRN would have to be printed on packs sold in Belgium. Assuming this is the case, and adding reciprocity, the pack could end up having six printed lines: batch number, expiry date, serial number, product code, and French and Belgian NHRNs. The more information to be printed, the more drastic the changes in the artwork could be and this may even impact pack size.
If member states require their individual NHRNs to be printed and costs are added to the printing process, companies might evaluate the profitability of their products for small markets and, in the worst-case scenario, potentially withdraw them. Such withdrawals could decrease access to affordable high-quality medicines and increase shortages of some products in some markets. Shortages is a challenge that particularly needs to be addressed and is, to a certain extent, already being tackled by the industry using multi-market packs. (To be clear – all examples in this article are fictitious and for illustrative purposes only.)
A solution proposed by the industry is for all member states to use the same coding scheme. Through the application of the same coding scheme, the use of a single serialised number would allow every user of the EMVS to identify its NHRN in the national repository to which it is linked. Belgian industry stakeholders are understood to be exploring this solution. Where the Belgian stakeholders currently are using the ‘CNK’ number; it is possible to organise a conversion table. This conversion table links the serialised codes to the CNK. This allows any pack sold on the Belgian market to be correctly identified for reimbursement purposes.
Pack coding guideline
EFPIA published its Pack Coding Guideline in 2008, in which it recommends the adoption of a single coding scheme to be used throughout Europe. This is a ‘living’ document that is updated whenever necessary. The EMVO stakeholders who represent the industry proposed an ‘EMVO Pack Coding Guideline’. After a first series of sessions where this option was discussed, non-industry representative associations indicated they could not participate and so the three industry associations continued with the aim of publishing an ‘Industry Pack Coding Guideline’. In this document, a recommended way forward is presented, ie, to adopt a unique standard for the coding of pharmaceutical products across Europe.
Several aspects were considered before the GS1 coding scheme was identified as a potential way forward. One aspect we would like to emphasise is that the GS1 standard allows an NHRN to be included for more than one country in one single code. In practice this would even mean that one code could identify the reimbursement status throughout Europe. A single code could therefore be used for reimbursement purposes in multiple countries
The future
The advantages of a single coding standard could include:
- It makes sense to use a global standard in a more globalised and harmonised world
- Impact and costs of changes to the pack design can be limited when applying a global standard
- Multi-country packs can still be used, without which access to some medicines could be limited and shortages could ensue
- When GS1 is being used, this does not prevent in any way the reimbursement policies of a country, not even for multi-market packs.
Biography
 JOHAN VERHAEGHE joined Medicines for Europe (formerly EGA) in 2015 as Falsified Medicines Directive Project Manager. In 2017, he took on additional responsibilities as National Policy Liaison. As coordinator of the National Associations Committee, Johan’s task is to manage the platform where different national associations not only share their ‘best practices and lessons learned’ but also discuss a broad variety of topics that are related to the industry.
JOHAN VERHAEGHE joined Medicines for Europe (formerly EGA) in 2015 as Falsified Medicines Directive Project Manager. In 2017, he took on additional responsibilities as National Policy Liaison. As coordinator of the National Associations Committee, Johan’s task is to manage the platform where different national associations not only share their ‘best practices and lessons learned’ but also discuss a broad variety of topics that are related to the industry.
Issue
Related topics
Packaging, Regulation & Legislation, Supply Chain, Therapeutics