Mass spectrometry in the development of protein biologics
Posted: 15 April 2017 | Ioannis A. Papayannopoulos (Celldex Therapeutics Inc.) | No comments yet
Biological products, usually referred to as biologics, constitute a significant segment of pharmaceuticals in development. Biologics include therapeutic proteins, antibody drugs, antibody-drug conjugates (ADCs), vaccines, carbohydrates drugs, blood and tissue-derived pharmaceuticals, gene therapies, even live cells and tissues…

Of these, protein therapeutics have been capturing an ever-increasing portion of drug sales. In 2015, seven of the top 10 prescription medications in terms of sales were protein pharmaceuticals, mainly antibodies; this trend is expected to continue. As new protein and antibody therapeutics are being developed, some older ones have lost or are about to lose patent protection and this, along with a streamlined regulatory environment, has given rise to the development of biosimilars, the equivalent of small-molecule drug generics to protein pharmaceuticals.
The development of protein drugs has been facilitated by advances in analytical technologies, which in turn have made it possible to examine and understand the structure, stability and behaviour of protein pharmaceuticals in ever-increasing detail, breadth and depth. Chief among these analytical technologies has been mass spectrometry (MS), typically coupled online with high-performance liquid chromatograph (HPLC). Mass spectrometric data are being used extensively to support regulatory filings for protein and antibody therapeutics.
Assessing proteins’ primary sequence by LC-MS
One of the earliest applications of mass spectrometry in the development of protein pharmaceuticals was the confirmation of the amino acid sequence of recombinant proteins. The basic approach has not changed much since the early days; however, analytical instrumentation technology has advanced significantly in the intervening years. Thus, liquid chromatography and mass spectrometry (LC-MS) instruments, and the software tools that have been developed and which are being continuously refined and upgraded, allow for in-depth characterisation of the sequence of proteins and antibodies.
Protein samples are digested with a proteolytic enzyme of known and reproducible specificity, typically after denaturation, reduction of disulphide bonds, and cysteine alkylation to ensure that the protein is unfolded and thus optimally accessible to the proteolytic enzyme. Trypsin (cleaves at the C-terminus of lysines and arginines), endoproteinase Lys-C (cleaves at the C-terminus of lysine), endoproteinase Asp-N (cleaves at the N-terminus of aspartic acid), and endoproteinase Glu-C (cleaves at the C-terminus of glutamic acid) are some of the enzymes used for LC-MS analysis of proteins and are commercially available from several different vendors.
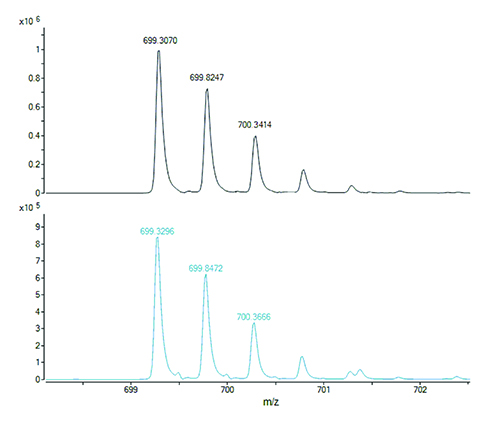
Figure 1. The doubly charged ions of two isobaric peptides, with different amino acid sequences, generated by digestion of a protein. The peptide with the all-12C containing isotope peak at m/z 699.3070 (top panel) has the molecular formula C62H88N14O21S1 and calculated m/z 699.3057 for its doubly charged ion (error 0.0013 u or 1.9 ppm). The peptide with the all-12C containing isotope peak at m/z 699.3296 (bottom panel) has the molecular formula C64H92N12O23 and calculated m/z 699.3272 for its doubly charged ion (error 0.0024 or 3.4 ppm). Cross assignment (ie, measured m/z 699.3070 to calculated m/z 699.3272 and measured m/z 699.3296 to calculated m/z 699.3057) would result in errors of 34 ppm and 29 ppm, respectively. Any ambiguities regarding the peptide assignments to the protein sequence can be resolved from the respective fragment ion mass spectra.
Proteolytic peptides thus generated are separated by HPLC (usually on reversed-phase columns) and their masses are measured with high accuracy (low parts-per-million) and can be matched to the calculated (theoretical) mass values of the expected peptides from a particular protein sequence. Peptide fragment ion mass spectra (MS/MS) are used to confirm the correctness of assigned masses to peptide sequences and to resolve ambiguities in the case of isomeric (same molecular formula hence same molecular mass) or isobaric peptides (molecular masses are almost the same and differ by an amount that may fall within the measurement error of the mass spectrometer, depending on the instrument used). The latter are illustrated in Figure 1.
In addition to establishing that the target amino acid sequence is as expected, LC-MS data of protein digests are also used to identify and confirm various modifications to the protein sequence. These include deamidation or oxidation of susceptible amino acids (eg, asparagines and methionines, respectively), pyroglutamate formation of N-terminus glutamines (frequently found on antibody-heavy chains), and truncation of the C-terminus. Many such modifications can be inferred by other analytical methods used on the intact protein. For example, differences in deamidation between samples of the same protein will affect the separation profiles in capillary isoelectric focusing or ion exchange chromatography. However, LC-MS analysis of proteolytic peptides allows the identification of the specific amino acids that are responsible for the profile changes in the other methods.
The likelihood that one or more such modifications may occur and the extent of these modifications are influenced by parameters such as pH, temperature and length of storage, and are closely related to the conditions of manufacturing (cell culture or fermentation) and purification, as well as the formulation buffers and excipients used. Mass spectrometry, therefore, has become an essential tool for assessing protein stability during development.
Protein carbohydrate analysis by MS
Another important consideration in establishing a reproducible manufacturing process, as well as in assessing stability, is protein glycosylation – different patterns of which have been shown to affect potency and immunogenicity of protein pharmaceuticals. LC-MS is used extensively in the characterisation of carbohydrates released from a protein either enzymatically (typically N-linked carbohydrates) or chemically (O-linked carbohydrates). Commercially available derivatisation reagents can be used to label the released carbohydrates so that they can be detected photometrically (ultraviolet or fluorescence detection) and also to improve their mass spectrometric response, so they can be measured accurately in the mass spectrometer as well. LC-MS of released and chemically labelled carbohydrates, along with the use of appropriate standards, can provide detailed information on their composition as well as sequence.
Ultimately, any carbohydrate composition or sequence information obtained by analysis of the released carbohydrate must be reconciled with the MS data obtained from intact protein molecular mass measurements or the measured mass of glycosylated peptides in enzymatic digests. For example, the difference between the measured molecular masses of intact glycosylated and deglycosylated protein can be used to confirm the presence of the carbohydrate expected to be on the protein. This is illustrated in Figure 2.
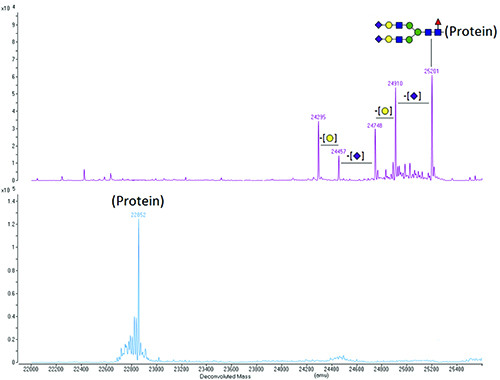
Figure 2. Deconvoluted molecular mass spectra of a glycosylated protein (top panel) and of the same protein after removal of the N-linked carbohydrate using the enzyme PNGase F (bottom panel). The species with the highest molecular mass (25201) is the protein with a fully sialylated core-fucosylated biantennary N-linked carbohydrate [(GlcNAc)4(Man)3(Gal)2(NeuAc)2(Fuc)1 – illustrated schematically on the top panel], which is typical for mammalian proteins. The heterogeneity of the glycosylated protein disappears upon deglycosylation, as it is due to the carbohydrate and not to the amino acid sequence.
Key: Purple rhombus – N-acetyl neuraminic acid (sialic acid, NeuAc); Yellow circle – Galactose (Gal); Blue square – N-acetyl glucosamine (GlcNAc); Green circle – Mannose (Man); Red triangle – Fucose (Fuc).
Protein disulphide characterisation
Disulphides play an important role in the folding of proteins and in maintaining their three- dimensional structure. The characterisation of disulphide bond connectivity can be complicated. This is especially true for proteins with many cysteines that can form disulphide bonds, such as antibodies (32 cysteines in IgG1, and 36 in IgG2). A typical workflow for the determination of disulphide connectivity would entail digestion of the antibody with one or more enzymes without prior reduction of cysteines, and assignment, often with the aid of specialised software, of disulphides from the MS data. However, conditions optimal for maintaining the integrity of disulphide bonds, such as low pH, are often suboptimal for digestion with the common proteolytic enzymes used in LC-MS analyses, and will vary from protein to protein.
The presence of reduced cysteines, a result of partially formed disulphide bonds between cysteines, has traditionally been assessed and quantitated colourimetrically with reagents that label free thiols. For small proteins or peptides with multiple cysteines the measured molecular mass can indicate whether a disulphide is present, as formation of a disulphide bond between two cysteines results in loss of 2u from the molecular mass (loss of two hydrogens). However, for larger proteins the mass accuracy and resolution of the mass spectrometer may be insufficient to determine definitively whether cysteines are present in reduced form or in disulphides. Labelling cysteines with different thiol-reactive molecules can be used to determine, from the increase in the measured molecular mass of a protein, the number of free thiols and the accessibility of cysteines and ease of reduction of specific disulphide bonds.
Assessing protein tertiary structure
Several physicochemical methods, such as x-ray crystallography, differential scanning calorimetry and spectroscopy, can provide useful information on the tertiary structure of proteins; x-ray crystallography in particular has been used to determine how a protein molecule presents in three dimensions. Hydrogen-deuterium exchange mass spectrometry (HDX-MS) is being used increasingly often to obtain similar information. The protein, antibody, or antibody fragment is incubated in deuterated water, resulting in the exchange of hydrogens of amide bonds with deuterons from the solvent. Since only hydrogens on amides that are exposed to the solvent are exchanged, the process is very sensitive to solvent accessibility and structure flexibility of the protein molecule.
The hydrogen-deuterium exchange is quenched by lowering the pH. This is followed by rapid digestion of the protein with pepsin and rapid LC-MS analysis of proteolytic peptides at low temperature to minimise the back exchange of deuterium with hydrogen. Automated liquid handling equipment that can operate at low temperatures ensures the standardisation of all steps critical for reproducibility.
The HDX-MS data are processed and presented in a readily interpretable format with specialised software. Peptides that exhibit significant deuterium incorporation are from regions of the protein sequence that are exposed to the solvent and peptides that exhibit little or none are from regions of the sequence that are shielded. Such data are correlated to protein folding and used to assess protein aggregation, and to compare different production batches or different antibodies of the same isotype such as biosimilars. Additionally, such data can be used for epitope mapping and ligand binding studies. HDX-MS has several advantages over x-ray crystallography as it requires significantly less material, is much faster and can tolerate impurities.
Analysis of protein biotherapeutics
Robust and accurate measurements (identification and quantitation) of protein biotherapeutics in complex matrices such as serum are essential in pharmacokinetic, pharmacodynamic and toxicokinetic studies during the development of biologics. Typically, this has been done using various ligand-binding assays; these are increasingly being supplemented and, in some cases, replaced by LC-MS methods. It is possible to measure intact protein in serum by MS, but most LC-MS assays of this type are carried out on peptides, generated by proteolysis. This is because MS sensitivity for smaller molecules such as peptides is much higher than for larger intact proteins, especially when utilising selected reaction monitoring (SRM) strategies instead of acquiring full mass spectra, an approach that has been used successfully for decades in similar analyses of small molecule drugs. Modifications can also complicate analysis of intact protein molecules, but peptides to be monitored can be selected from regions which are known not to be modified. Such LC-MS approaches typically offer better specificity, faster method development and validation, and lower sample consumption compared to ligand binding assays and are also more easily transferable between different animal models and to human samples.
Tissue imaging
Tissue imaging with mass spectrometry has been in use for some time to monitor the distribution, with very good spatial resolution, of small molecule biomarkers or therapeutics in tissues samples. An excised tissue slice is “scanned” down and across with a laser or other ion desorption method and the ions thus generated are analysed with a mass spectrometer. Ion abundances can be “mapped” across the tissue, thus enabling the measurement of concentration gradients of molecules of interest.
Intact proteins, however, ionise with low efficiency in tissue samples and any therapeutic protein or antibody signals would be interfered with or obscured by those of the abundant endogenous proteins. Digestion to generate smaller peptides cannot be carried out effectively on a tissue without losing localisation of the protein of interest. However, with ADCs it is the distribution of the “payload” – the small molecule chemotherapeutic drug – covalently attached to the antibody that delivers it to the appropriate location in the body (eg, the site of a tumour), which is primarily of interest. Using laser desorption as the ionisation method causes the bond between the small molecule drug and the antibody to break, thus enabling the former to be ionised and detected. This approach has the advantage of being faster than immunohistochemistry to develop, validate and use routinely.
Conclusion
The scope and applications of mass spectrometric analyses continue to expand and encompass all aspects of protein characterisation (primary structure, modifications, folding, binding), as well as metabolism and pharmacokinetic and pharmacodynamic applications. Mass spectrometry coupled online to liquid chromatography has played a critical role in the development of biopharmaceuticals since the early days of the biotechnology industry and undoubtedly will continue to do so in the future.
Biography
 IOANNIS PAPAYANNOPOULOS has been carrying out peptide and protein analysis and characterisation work, using mass spectrometry, chromatography and other analytical techniques, for over a quarter century. He received his undergraduate degree in chemistry from Bowdoin College and his PhD in organic chemistry from the Massachusetts Institute of Technology, where he conducted research in mass spectrometry under the supervision of the late Klaus Biemann. He has held senior scientific and management positions in the biopharmaceutical industry, at such companies as AstraZeneca, Biogen, EMD Pharmaceuticals and Targanox, and in academia, most recently as the director of the proteomics core facility at the Koch Institute for Integrative Cancer Research at MIT. Currently he is working on the analysis and characterisation of antibodies, antibody-drug conjugates, and recombinant protein pharmaceuticals at Celldex Therapeutics, a biotechnology company in Massachusetts.
IOANNIS PAPAYANNOPOULOS has been carrying out peptide and protein analysis and characterisation work, using mass spectrometry, chromatography and other analytical techniques, for over a quarter century. He received his undergraduate degree in chemistry from Bowdoin College and his PhD in organic chemistry from the Massachusetts Institute of Technology, where he conducted research in mass spectrometry under the supervision of the late Klaus Biemann. He has held senior scientific and management positions in the biopharmaceutical industry, at such companies as AstraZeneca, Biogen, EMD Pharmaceuticals and Targanox, and in academia, most recently as the director of the proteomics core facility at the Koch Institute for Integrative Cancer Research at MIT. Currently he is working on the analysis and characterisation of antibodies, antibody-drug conjugates, and recombinant protein pharmaceuticals at Celldex Therapeutics, a biotechnology company in Massachusetts.
References
- US Food and Drug Administration. What are “Biologics” Questions and Answers.
https://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CBER/ucm133077.htm - Top drugs by sales revenue in 2015: Who sold the biggest blockbuster drugs?
http://www.pharmacompass.com/radio-compass-blog/top-drugs-by-sales-revenue-in-2015-who-sold-the-biggest-blockbuster-drugs - Beck A, Sanglier-Cianferani S, Van Dorsselaer A. Biosimilar, Biobetter, and Next Generation Antibody Characterization by Mass Spectrometry. Analytical Chemistry. 2013;84:4637-4646.
- Trouvin J-H. Biosimilar medicinal products – EU experience and perspectives.
http://www.diaglobal.org/productfiles/24338/5%203.prof%20jean-hugues%20trouvin.pdf. - Rogstad S, Faustino A, et al. A Retrospective Evaluation of the Use of Mass Spectrometry in FDA Biologics License Applications. Journal of the American Society for Mass Spectrometry. published online: November 21, 2016.
- Hmiel LK, Brorson KA, Boyne MT 2nd. Post-translational structural modifications of immunoglobulin G and their effect on biological activity. Analytical and Bioanalytical Chemistry. 2015;407:79–94.
- Papayannopoulos IA. The interpretation of collision-induced dissociation tandem mass spectra of peptides. Mass Spectrometry Reviews 1995;14:49–73.
- Hermeling S, Crommelin DJ, et al. Structure–immunogenicity relationships of therapeutic proteins. Pharmaceutical Research 2004;21:897–903.
- An B, Zhang M, and Qu J. Toward Sensitive and Accurate Analysis of Antibody Biotherapeutics by Liquid Chromatography Coupled with Mass Spectrometry. Drug Metabolism and Disposition. 2014;42:1858-1866.
- Jujiwara Y, Furuta M, et al. Imaging mass spectrometry for the precise design of antibody-drug conjugates. Nature Scientific Reports 6, published online: April 21, 2016.