Rational drug design: How far have we come?
Posted: 8 January 2018 | Simon Pearce ( Thermo Fisher Scientific) | No comments yet
From the early use of Hansch parameters and Topliss Trees to today’s computational structure–activity techniques, medicinal chemists have long sought to rationalise drug design to find the quickest and most resource-efficient route to market. But while modern strategies are more reliant on statistical algorithms and vast data libraries, these founding principles remain at their core…

Medicinal chemists may spend years tweaking the structure of initial candidate molecules in order to improve their potency, toxicology and pharmacokinetic profile. But while the molecular ‘hits’ identified during the discovery stage are a good place to start drug development, they’re usually some degree off the ideal structure of a finished product.
With aromatic substructures highly prevalent in pharmaceutical products, the focus is often on modifying the substitution patterns of phenyl rings. However, with a wealth of substituents to choose from, and a seemingly unlimited choice of configurations, synthesizing and analysing the activities of each structure would be a very slow and costly option. When it comes to drug development, a rational approach is required.
Rational beginnings
Early drug development was predominantly a trial-and-error process, with structure optimisation guided largely by the intuition of medicinal chemists and the availability of starting materials.
However, in the mid-1960s Corwin Hansch pioneered a more systematic approach to the design of aromatic substructures, built on knowledge of how a drug molecule’s biological activity correlated with the properties of its phenyl ring substituents1. This method was an early form of what we now know as a quantitative structure-activity relationship (QSAR) study.
Through careful analysis, Hansch identified that pharmacological activity was often influenced by physical factors such as the electron withdrawing or donating characteristics of ring substituents (Hammett substituent coefficient σ), their relative hydrophobicity (π constant), as well as their size and shape (Taft steric factor Es).
By studying the relationship between these physicochemical properties and the biological activity of the molecule, novel analogues could be designed with the appropriate functionality. Moreover, if a molecule’s activity was not found to correlate with σ, π or Es properties, this suggested an as yet unidentified structural characteristic had an important part to play in dictating its biological behaviour.
Branching out
Despite the logic behind this approach, few medicinal chemists actually put Hansch’s methods into practice. To obtain the most reliable correlations, a relatively large number of molecules had to be used; yet preparing a large set of structurally analogous molecules could be time-consuming and synthetically challenging. Other limitations, such as the variability of binding modes between compounds, the effects of alkyl chains, as well as some chemists’ aversion to adopt mathematical drug design approaches, also contributed to this poor uptake.
In an effort to overcome these challenges, in 1972 John Topliss published a non-mathematical strategy for the design of drug molecules2, based on the underlying physicochemical properties studied by Hansch. This more ‘user-friendly’ approach required chemists to make a series of stepwise decisions based on the relative potencies of the compounds prepared so far. Depending on whether a molecule was more, less or equally as potent as the molecule previously produced, the decision tree would guide the chemist towards the next structure to synthesize and investigate (Figure 1).
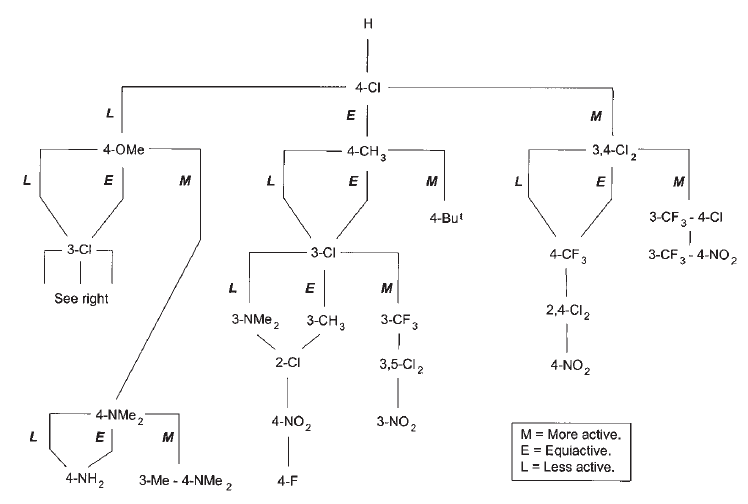
Figure 1. Topliss decision tree for aromatic substituents
The Topliss Tree, as it came to be known, was more widely adopted by medicinal chemists and helped to make drug design considerably more efficient, particularly in the early stages of structure optimisation.
Never far from the tree?
In the four decades since the publication of the Topliss Tree strategy, advances in the capability of statistical analysis software, as well as an expansion of physical and biological data associated with drug molecule potency, have greatly enhanced the potential of QSAR approaches for drug design.
The rest of this content is restricted - login or subscribe free to access
 Thank you for visiting our website. To access this content in full you'll need to login. It's completely free to subscribe, and in less than a minute you can continue reading. If you've already subscribed, great - just login.
Thank you for visiting our website. To access this content in full you'll need to login. It's completely free to subscribe, and in less than a minute you can continue reading. If you've already subscribed, great - just login.
Why subscribe? Join our growing community of thousands of industry professionals and gain access to:
- bi-monthly issues in print and/or digital format
- case studies, whitepapers, webinars and industry-leading content
- breaking news and features
- our extensive online archive of thousands of articles and years of past issues
- ...And it's all free!
Click here to Subscribe today Login here