Glucosamine is sold as a food supplement worldwide, but its production is poorly regulated. A direct and reliable method for the quality control of glucosamine in commercial products is of great importance. This article describes a simple, fast and validated ultra-performance liquid chromatography-quadrupole time of flight (UPLC-QToF) for the determination of glucosamine in commercial food supplements.
Glucosamine(2-amino-2-deoxy-dglucose), deriving from the natural amino monosaccharide, is an essential component of glycosaminoglycans in the articular cartilage and synovial fluid.1 Numerous clinical trials and in vitro studies have demonstrated that exogenous administration of glucosamine can relieve osteoarthritis (OA) symptoms and restore articular functions by stimulating the synthesis of proteoglycans and protecting articular cartilage. It also has considerable anti-inflammatory and antioxidative effects.2-4 In addition, it is reported that glucosamine has no major adverse effects and can be tolerated well by humans because of its natural occurrence in the body.5,6
This webinar showcases the Growth Direct System; an RMM (Rapid Microbial Method) that improves on traditional membrane filtration, delivering increased accuracy, a faster time to result, enhanced data integrity compliance, and more control over the manufacturing process.
Key learning points:
Understand the benefits of full workflow microbiology quality control testing automation in radiopharmaceutical production
Learn about ITM’s implementation journey and considerations when evaluating the technology
Find out how the advanced optics and microcolony detection capabilities of Growth Direct® technology impact time to result (TTR).
Don’t miss your chance to learn from experts in the industry –Register for FREE
Can’t attend live? No worries – register to receive the recording post-event.
Therefore, glucosamine is recommended for the management of OA by several guidelines from international societies and agencies like the Osteoarthritis Research Society International (OARSI), which suggests glucosamine as a symptom-relieving and structure-modifying agent for OA.7 The European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO) recommended glucosamine sulfate as the first-line pharmacological treatment for slow-onset medium to long-term control of symptoms.8 The European League Against Rheumatism (EULAR) guidelines specified glucosamine as, “highest level of evidence and strength of recommendation” for the treatment of knee OA.9 The United States Food and Drug Administration (FDA) considers glucosamine as a dietary supplement and states that there is no sufficient evidence for any medicinal effects.10
At present, glucosamine products are one of the most popular over-the-counter (OTC) food supplements and are widely used all over the world. Glucosamine can be taken as a single supplement in the forms of glucosamine hydrochloride (GlcN•HCl), glucosamine sulphate ((GlcN)2•H2SO4), and glucosamine sulphate 2KCl ((GlcN)2•H2SO4•2KCl). Alternatively, it can be combined with other nutraceutical supplements such as chondroitin sulphate. Given that there are a large variety of glucosamine products available in the market and the current quality control of dietary supplements lacks regulatory provisions,11 there is a need to develop simple methods to determine amounts of glucosamine in these products.
The main challenges in analysing glucosamine are the lack of UV absorption chromophore and its high polarity.12 Glucosamine is usually analysed by reverse-phase high-performance liquid chromatography with ultraviolet detection (RP-HPLC-UV), which is by far the most widely applied for routine analysis.13 HPLC coupled to other detection techniques like electrochemical detection (ECD),14 refractive index detection (RI)15 and evaporative light scattering detection (ELSD)16 have also been reported, to bypass poor UV absorption. To overcome short retention issues, HPLC-UV with pre-column derivatisation was developed for the analysis of glucosamine to enforce detectability and achieve sufficient retention on reverse phase columns.17-19 Yet again, derivatisation can be a time-consuming procedure for sample preparation and HPLC methods with mass spectrometry (MS) or tandem mass spectrometry (MS/MS) for direct analysis of glucosamine from plasma have been achieved with normal phase columns, like amino20 and cyano columns,21 which obtained both shorter analytical time and sensitivity of glucosamine.
Hydrophilic interaction liquid chromatography (HILIC) applies a hydrophilic stationary phase and aqueous-organic mobile phase and has proven to be an ideal tool for the analysis of polar compounds that are poorly retained on reversed phase columns22,23 and it is highly compatible with LC-MS.24,25 HILIC-ESI-MS has already been used to study carbohydrates,26,27 and applied to determine glucosamine in different matrices such as tobacco,28 fungal biomass29 and dog plasma.12
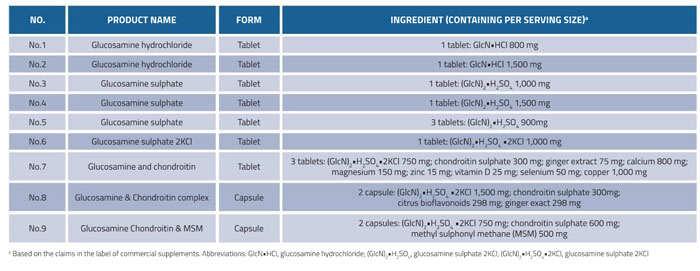
Here we develop an UPLC-QToF-MS method with a ZIC-HILIC column for the determination of glucosamine in nine representative dietary supplements from different brands and manufacturers. Table 1 shows the label information for each type of supplement.
Table 1: Information from nine batches of commercial glucosamine-containing supplements
Optimising sample preparation and LC/MS conditions
The extractant and ultrasonication time were optimised for the sample preparation procedure. Glucosamine is an amino sugar which is freely soluble in water. Based on its dissolution characteristics, several reports used water as the extractant of glucosamine.30,31 The United States Pharmacopoeia 36 (USP36) applies 50% acetonitrile and 50% water (v/v) as the extractant. Herein the extraction efficiency of these two solvents was compared using peak area of glucosamine and there was no difference in extraction efficiency between water and 50% acetonitrile. Moreover, when using HILIC it is recommended that sample solvents should consist of 50-100% organic solvent. Therefore, 50% acetonitrile and 50% water (v/v) was chosen as the extractant. The ultrasonication time for sample extraction was optimised to get the optimal extraction efficiency. Peak area was used to investigate the effect of different ultrasonication times (10, 20, and 30min). The results showed that peak area of glucosamine increased from 10min to 20min, but it no longer increased as the time lengthened. Thus, 20min was selected as the ultrasonication time for sample extraction.
In HILIC, ammonium formate or ammonium acetate buffer in the concentration range 5-20mM is recommended for most analytes and acetonitrile is suitable for HILIC separations. Based on the above, mobile phase consisting of acetonitrile: 10mM ammonium formate (60:40) was assessed.
A broad and tailed peak was obtained for the glucosamine analyte. When gradient elution was employed, peak tailing was not improved. When the pH of the mobile phase was adjusted to 3.0 with formic acid, the peak was improved. To ensure pH consistency formic acid and ammonium formate were added to both solvents, water containing 0.1% formic acid and 10mM ammonium formate (mobile phase A) and 80% acetonitrile containing 0.1% formic acid and 10mM ammonium formate (mobile phase B) were used as the mobile phases.
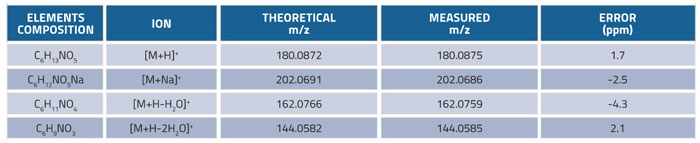
Table 2: Calculated and measured molecular weights of glucosamine and its fragment ions
Flow injection analysis (FIA) experiments using a standard solution of glucosamine at a concentration of 25μg/mL were used to optimise the MS parameters. The protonated molecular ion peak [M+H]+ was selected as the quantitative ion and monitored at m/z: 180.0875. The electrospray ionisation source in positive mode was set with collision energy (CE) of 4eV and the capillary and cone voltages were set at 3.2kV and 45V respectively. Mass fragmentation pattern and characteristic fragment ions of glucosamine were consistent with the findings reported in previous literature.29 The calculated molecular weight and measured molecular weight of glucosamine and its fragment ions are shown in Table 2. The errors against the theoretical value were below 5ppm, indicating optimum accuracy for the Q-TOF mass detector.
Detection, precision and recovery
The linearity was evaluated by analysing six standard solutions in the range of 12.5-400μg/mL. Each standard was analysed in triplicate. The linear regression analysis for glucosamine hydrochloride was constructed by plotting the peak area against concentration. The linear regression equation for calibration curve was Y=19.12X+338.15 with correlation coefficient r2 = 0.9969, indicating good linearity. Limit of detection (LOD) and limit of quantitation (LOQ) were measured as the concentrations corresponding to the signal-to-noise ratio of 3:1 and 10:1, respectively. The LOD and LOQ values for glucosamine were found to be 0.25 and 1.0μg/mL, respectively.
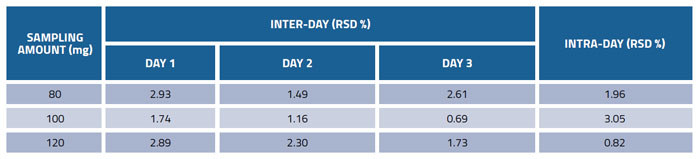
Table 3: Precision of the method for determination of glucosamine in dietary supplements
The precision of the method was demonstrated by analysing commercial product (No.1) at three different sampling amounts (80, 100, 120 mg) in triplicates on three consecutive days. The results in Table 3 show that the values of precision for inter-day variations were between 0.69 and 2.93%, and intra-day variations were between 0.82 and 3.05%, indicating excellent precision.
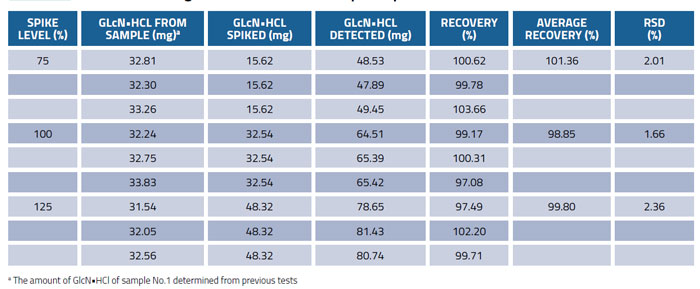
The recovery test was carried out to further investigate the accuracy of the method by spiking glucosamine hydrochloride standard solution at 75, 100 and 125% of the expected concentrations to the known amount of commercial product sample (No.1). The samples were prepared in triplicates at each level. The recovery results presented in Table 4 demonstrate both good reproducibility and accuracy. The average recoveries were in the range of 98.85-101.36% and the RSD was ≤3.0%, which proved the reliability of this method.
Table 4: Recoveries of glucosamine from samples spiked at different levels (n=3)
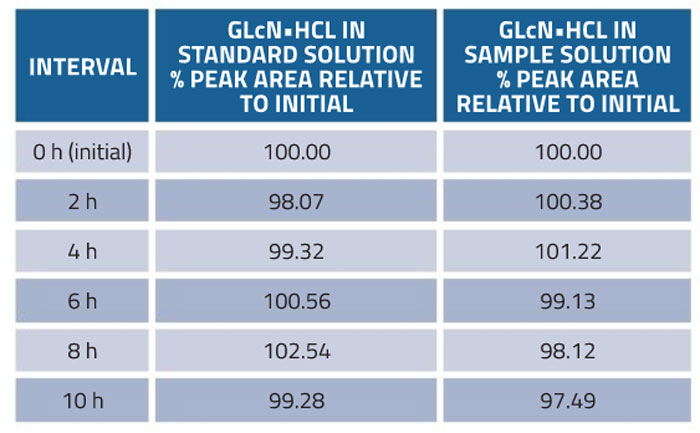
Stability of glucosamine in the standard and sample was assessed at room temperature. The solutions were analysed in triplicate every 2h within 10h. The results in Table 5show that standard and sample solutions retained a potency of 100.0 ±2.0% as compared with the fresh solution over the tested period.
Table 5: Stability of the method for determination of glucosamine in dietary supplements
Determination of glucosamine in dietary supplements
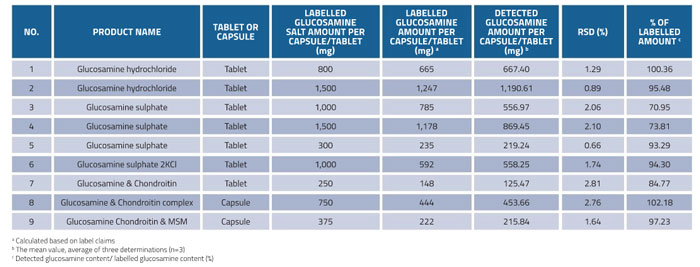
In order to evaluate the quality of glucosamine supplements, the developed HILIC-ESI-MS method was applied for the determination of glucosamine in nine samples of marketed dietary supplements (No.1-No.9). As can be seen from Table 6, the amounts of glucosamine in commercial supplements were 125.47-1,190.61mg/tablet or mg/capsule, and the results showed good reproducibility with RSD values of less than 3%. The ratio of detected glucosamine contents to labelled amount ranged from 70.95 to 102.18%. Three out of nine products were found to contain less than 10% of the amount of glucosamine claimed on the label. Two products contained less than 20% of the stated amount (70.95% and 73.81% for No.3 and No.4, respectively). Most of the products (six out of nine) met the label claim, which satisfied the industry-accepted 10% deviation from the stated amount (USP 36).
Table 6: Determination of glucosamine contents in commercial dietary supplements
HPLC analysis of glucosamine in dietary supplements is performed with an amino column in USP 36, while an ion-pair reversed-phase HPLC method is applied by the British Pharmacopoeia (BP) 2015. Since glucosamine has no UV absorption except in the region of very short wavelength (end absorption), both official methods are carried out at the detection wavelength of 195nm and this can result in high noise in the baseline, which can, in turn, affect sensitivity. The AOAC International has established an official method for determination of glucosamine in dietary supplements by HPLC with FMOC-Su derivatisation,32 but the implementation of this method can be time-consuming and increase the cost of analysis.
This work aimed at assessing glucosamine by hydrophilic interaction liquid chromatography coupled to electron spray ionisation mass spectrometry for the quality control of glucosamine products. The method used a ZIC-HILIC column coupled to mass spectrometry in the positive mode and applied for the determination of glucosamine content commercially available capsules or tablets of glucosamine. HILIC was the column of choice due to previous reports where C18 or C8 columns were employed and the authors reported that glucosamine and the salt ions, such as chloride or sulphate, in food supplements could not be separated – their peaks almost completely overlapped in chromatograms (Zheng, Li, Wang and Wang, 2008).
According to the USP, glucosamine supplements should contain 90-110% of the label claim. The results of this investigation demonstrated that 33% of products failed to meet the acceptance criteria of the USP (less than 10% of the stated amount). One explanation for the variability in glucosamine levels is that manufacturers do not need to undergo a strict quality control process like those followed by drugs. Based on the discussion above, there is an urgent need for manufacturers of glucosamine supplements to improve quality control methods.
Conclusion
In this work, a simple, sensitive and reliable HILIC-ESI-MS method for the direct quantitative determination of glucosamine in commercial food supplements without the need for pre-analytical derivatisation has been developed. It was validated with high accuracy and precision. This approach is an alternative to the previous methods in terms of quality control of glucosamine food supplements.
References
Dostrovsky N, Towheed T, Hudson R. Anastassiades T. The effect of glucosamine on glucose metabolism in humans: a systematic review of the literature. Osteoarthritis and Cartilage. 2011;19(4):375-380.
Dalirfardouei R, Karimi G, Jamialahmadi K. Molecular mechanisms and biomedical applications of glucosamine as a potential multifunctional therapeutic agent. Life sciences. 2016;152:21-29.
Chan KOW, Ng GYF. A review on the effects of glucosamine for knee osteoarthritis based on human and animal studies. Hong Kong Physiotherapy Journal. 2011;29(2):42-52.
Nakamura H. Application of glucosamine on human disease—osteoarthritis Carbohydrate polymers. 2011;84(2):835-839.
Bruyère O, Altman RD. Reginster J-Y. Efficacy and safety of glucosamine sulfate in the management of osteoarthritis: evidence from real-life setting trials and surveys. Seminars in arthritis and rheumatism. Elsevier. 2016; pp.S12-S17.
Rovati LC, Girolami F, Persiani S. Crystalline glucosamine sulfate in the management of knee osteoarthritis: efficacy, safety, and pharmacokinetic properties. Therapeutic advances in musculoskeletal disease. 2012. 1759720X12437753.
Zhang W, Moskowitz R, Nuki G, Abramson S, Altman R, Arden N, Bierma-Zeinstra S, Brandt K, Croft P, Doherty M. OARSI recommendations for the management of hip and knee osteoarthritis, Part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis and cartilage. 2008;16(2):137-162.
Bruyère O, Cooper C, Pelletier J-P, Branco J, Brandi ML, Guillemin F, Hochberg MC, Kanis JA, Kvien TK, Martel-Pelletier J. An algorithm recommendation for the management of knee osteoarthritis in Europe and internationally: a report from a task force of the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO). Seminars in arthritis and rheumatism. Elsevier. 2014. 253-263.
Jordan K, Arden N, Doherty M, Bannwarth B, Bijlsma J, Dieppe P, Gunther K, Hauselmann H, Herrero-Beaumont G, Kaklamanis P. EULAR Recommendations 2003: an evidence based approach to the management of knee osteoarthritis: Report of a Task Force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Annals of the rheumatic diseases. 2003;62(12):1145-1155.
Mantovani V, Maccari F, Volpi N. Chondroitin Sulfate and Glucosamine as Disease Modifying Anti-Osteoarthritis Drugs (DMOADs). Current medicinal chemistry. 2016;23(11):1139-1151.
Aghazadeh-Habashi A, Duke J, Jamali F. The Impact of Implementation of the Canadian Regulatory Requirements on the Quality of Natural Health Products: The Glucosamine Case, Journal of Pharmacy & Pharmaceutical Sciences. 2014;17(1):20-24.
Hubert C, Houari S, Lecomte F, Houbart V, De Bleye C, Fillet M, Piel G, Rozet E, Hubert P. Development and validation of a sensitive solid phase extraction/hydrophilic interaction liquid chromatography/mass spectrometry method for the accurate determination of glucosamine in dog plasma. Journal of Chromatography A. 2010;1217(19):3275-3281.
Akamatsu S, Mitsuhashi T. Development of a simple capillary electrophoretic determination of glucosamine in nutritional supplements using in-capillary derivatisation with o-phthalaldehyde. Food Chemistry. 2012;130(4):1137-1141.
Pashkova E, Pirogov A, Bendryshev A, Ivanaynen E, Shpigun O. Determination of underivatized glucosamine in human plasma by high-performance liquid chromatography with electrochemical detection: Application to pharmacokinetic study. Journal of pharmaceutical and biomedical analysis. 2009;50(4):671-674.
Xinmin W, Ruili Z, Zhihua L, Yuanhong W, Tingfu J. Determination of glucosamine and lactose in milk-based formulae by high-performance liquid chromatography. Journal of food composition and analysis. 2008; 21(3):255-258.
Kim HJ, Bae IK, Jeong MH, Park HJ, Jung JS, Kim JE. A New HPLC-ELSD Method for Simultaneous Determination of N-Acetylglucosamine and N-Acetylgalactosamine in Dairy Foods. International journal of analytical chemistry. 2015.
Su G, Wang X, Chi D, Li L, Shao L. Improved Determination of d‐Glucosamine Hydrochloride in Health Foods by HPLC Using 7‐Fluoro‐4‐Nitrobenzo‐2‐Oxa‐l, 3‐Diazole as a Derivative. Journal of food science. 2011;76(9):N74-N78.
Hadad GM, Abdel-Salam RA, Emara S. Determination of Glucosamine and Carisoprodol in pharmaceutical formulations by LC with pre-column derivatization and UV detection. Journal of chromatographic science. 2012 bms008.
Sanches‐Silva A, Ribeiro T, Albuquerque TG, Paseiro P, Sendón R, de Quirós AB, López‐Cervantes J, Sánchez‐Machado DI, Valdez HS, Angulo I. Ultra‐high pressure LC determination of glucosamine in shrimp by‐products and migration tests of chitosan films. Journal of separation science. 2012;35(5-6):633-640.
Roda A, Sabatini L, Barbieri A, Guardigli M, Locatelli M, Violante FS, Rovati LC, Persiani S. Development and validation of a sensitive HPLC–ESI-MS/MS method for the direct determination of glucosamine in human plasma. Journal of Chromatography B. 2006;844(1):119-126.
Zhong S, Zhong D, Chen X. Improved and simplified liquid chromatography/electrospray ionization mass spectrometry method for the analysis of underivatized glucosamine in human plasma. Journal of Chromatography B. 2007;854(1):291-298.
Liu M, Chen EX, Ji R, Semin D. Stability-indicating hydrophilic interaction liquid chromatography method for highly polar and basic compounds. Journal of chromatography A. 2008;1188(2):255-263.
Gika HG, Theodoridis GA, Vrhovsek, U, Mattivi F. Quantitative profiling of polar primary metabolites using hydrophilic interaction ultrahigh performance liquid chromatography–tandem mass spectrometry. Journal of Chromatography A. 2012; 1259:121-127.
Mitchell CR, Bao Y, Benz NJ, Zhang S. Comparison of the sensitivity of evaporative universal detectors and LC/MS in the HILIC and the reversed-phase HPLC modes. Journal of Chromatography B. 2009;877(32):4133-4139.
Zhang T, Creek DJ, Barrett MP, Blackburn G, Watson DG. Evaluation of coupling reversed phase, aqueous normal phase, and hydrophilic interaction liquid chromatography with Orbitrap mass spectrometry for metabolomic studies of human urine. Analytical chemistry; 2012;84(4):1994-2001.
Hernández-Hernández O, Calvillo I, Lebrón-Aguilar R, Moreno FJ, Sanz ML. Hydrophilic interaction liquid chromatography coupled to mass spectrometry for the characterization of prebiotic galactooligosaccharides. Journal of Chromatography A. 2012;1220:57-67.
Rodriguez-Sanchez S, Quintanilla-López JE, Soria AC, Sanz M. Evaluation of different hydrophilic stationary phases for the simultaneous determination of iminosugars and other low molecular weight carbohydrates in vegetable extracts by liquid chromatography tandem mass spectrometry. Journal of Chromatography A. 2014;1372:81-90.
Moldoveanu SC, Byrd CH, Gerardi AR. Analysis of certain nitrogenous compounds in tobacco. Part 1: adenosine, 2, 5-and 2, 6-deoxyfructosazines, mannosamine and glucosamine. Beiträge zur Tabakforschung/Contributions to Tobacco Research. 2001:24(5):234-242.
Olofsson MA, Bylund D. Liquid Chromatography with Electrospray Ionization and Tandem Mass Spectrometry Applied in the Quantitative Analysis of Chitin-Derived Glucosamine for a Rapid Estimation of Fungal Biomass in Soil. International Journal of Analytical Chemistry. 2016.
Shen X, Yang M, Tomellini SA. Liquid chromatographic analysis of glucosamine in commercial dietary supplements using indirect fluorescence detection. Journal of chromatographic science. 2007;45(2):70-75.
Magaña AA, Wrobel K, Escobosa ARC, Wrobel K. Fast determination of glucosamine in pharmaceutical formulations by high performance liquid chromatography without pre-column derivatization. Acta Universitaria. 2014;24(2):16-22.
Zhou JZ, Waszkuc T, Mohammed F. Determination of glucosamine in raw materials and dietary supplements containing glucosamine sulfate and/or glucosamine hydrochloride by high-performance liquid chromatography with FMOC-Su derivatization: Collaborative study. Journal of AOAC International. 2005;88(4):1048-1058.
Author biographies
DR CRISTINA LEGIDO-QUIGLEY – is an Associate Professor and Senior Lecturer in the IPS, Kings College London and works in the chemistry and medicine interface. She is an expert in analytical methodology and particularly in metabolomics/ lipidomics. To date she has taught analytical chemistry to 2,500+ students.
LIN ZHENG is a Professor at Guizhou Medical University of China and worked as an academic visitor at Dr Cristina Legido-Quigley’s group at King’s College London in 2016. Lin focuses on pharmaceutical analysis and quality control for Chinese herbal medicines and is in charge of two projects for the National Natural Science Foundation of China.
PEI HAN is a current PhD student in analytical chemistry at King’s College London, under the supervision of Dr Cristina Legido-Quigley. She studied traditional Chinese medicine for her first degree and now focuses on the application of metabolomics in Chinese herbal medicine research.
MIN KIM is a final year PhD student at King’s College London. His research/ PhD is on the identification of Alzheimer’s disease biomarkers using metabolomics. To date, he has analysed over 1,500 human blood samples using LC-MS, which has resulted in six publications of which he is the first co-author of two.
This website uses cookies to enable, optimise and analyse site operations, as well as to provide personalised content and allow you to connect to social media. By clicking "I agree" you consent to the use of cookies for non-essential functions and the related processing of personal data. You can adjust your cookie and associated data processing preferences at any time via our "Cookie Settings". Please view our Cookie Policy to learn more about the use of cookies on our website.
This website uses cookies to improve your experience while you navigate through the website. Out of these cookies, the cookies that are categorised as ”Necessary” are stored on your browser as they are as essential for the working of basic functionalities of the website. For our other types of cookies “Advertising & Targeting”, “Analytics” and “Performance”, these help us analyse and understand how you use this website. These cookies will be stored in your browser only with your consent. You also have the option to opt-out of these different types of cookies. But opting out of some of these cookies may have an effect on your browsing experience. You can adjust the available sliders to ‘Enabled’ or ‘Disabled’, then click ‘Save and Accept’. View our Cookie Policy page.
Necessary cookies are absolutely essential for the website to function properly. This category only includes cookies that ensures basic functionalities and security features of the website. These cookies do not store any personal information.
Cookie
Description
cookielawinfo-checkbox-advertising-targeting
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertising & Targeting".
cookielawinfo-checkbox-analytics
This cookie is set by GDPR Cookie Consent WordPress Plugin. The cookie is used to remember the user consent for the cookies under the category "Analytics".
cookielawinfo-checkbox-necessary
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
This cookie is set by GDPR Cookie Consent WordPress Plugin. The cookie is used to remember the user consent for the cookies under the category "Performance".
PHPSESSID
This cookie is native to PHP applications. The cookie is used to store and identify a users' unique session ID for the purpose of managing user session on the website. The cookie is a session cookies and is deleted when all the browser windows are closed.
viewed_cookie_policy
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
zmember_logged
This session cookie is served by our membership/subscription system and controls whether you are able to see content which is only available to logged in users.
Performance cookies are includes cookies that deliver enhanced functionalities of the website, such as caching. These cookies do not store any personal information.
Cookie
Description
cf_ob_info
This cookie is set by Cloudflare content delivery network and, in conjunction with the cookie 'cf_use_ob', is used to determine whether it should continue serving “Always Online” until the cookie expires.
cf_use_ob
This cookie is set by Cloudflare content delivery network and is used to determine whether it should continue serving “Always Online” until the cookie expires.
free_subscription_only
This session cookie is served by our membership/subscription system and controls which types of content you are able to access.
ls_smartpush
This cookie is set by Litespeed Server and allows the server to store settings to help improve performance of the site.
one_signal_sdk_db
This cookie is set by OneSignal push notifications and is used for storing user preferences in connection with their notification permission status.
YSC
This cookie is set by Youtube and is used to track the views of embedded videos.
Analytics cookies collect information about your use of the content, and in combination with previously collected information, are used to measure, understand, and report on your usage of this website.
Cookie
Description
bcookie
This cookie is set by LinkedIn. The purpose of the cookie is to enable LinkedIn functionalities on the page.
GPS
This cookie is set by YouTube and registers a unique ID for tracking users based on their geographical location
lang
This cookie is set by LinkedIn and is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
lidc
This cookie is set by LinkedIn and used for routing.
lissc
This cookie is set by LinkedIn share Buttons and ad tags.
vuid
We embed videos from our official Vimeo channel. When you press play, Vimeo will drop third party cookies to enable the video to play and to see how long a viewer has watched the video. This cookie does not track individuals.
wow.anonymousId
This cookie is set by Spotler and tracks an anonymous visitor ID.
wow.schedule
This cookie is set by Spotler and enables it to track the Load Balance Session Queue.
wow.session
This cookie is set by Spotler to track the Internet Information Services (IIS) session state.
wow.utmvalues
This cookie is set by Spotler and stores the UTM values for the session. UTM values are specific text strings that are appended to URLs that allow Communigator to track the URLs and the UTM values when they get clicked on.
_ga
This cookie is set by Google Analytics and is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. It stores information anonymously and assign a randomly generated number to identify unique visitors.
_gat
This cookies is set by Google Universal Analytics to throttle the request rate to limit the collection of data on high traffic sites.
_gid
This cookie is set by Google Analytics and is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visited in an anonymous form.
Advertising and targeting cookies help us provide our visitors with relevant ads and marketing campaigns.
Cookie
Description
advanced_ads_browser_width
This cookie is set by Advanced Ads and measures the browser width.
advanced_ads_page_impressions
This cookie is set by Advanced Ads and measures the number of previous page impressions.
advanced_ads_pro_server_info
This cookie is set by Advanced Ads and sets geo-location, user role and user capabilities. It is used by cache busting in Advanced Ads Pro when the appropriate visitor conditions are used.
advanced_ads_pro_visitor_referrer
This cookie is set by Advanced Ads and sets the referrer URL.
bscookie
This cookie is a browser ID cookie set by LinkedIn share Buttons and ad tags.
IDE
This cookie is set by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
li_sugr
This cookie is set by LinkedIn and is used for tracking.
UserMatchHistory
This cookie is set by Linkedin and is used to track visitors on multiple websites, in order to present relevant advertisement based on the visitor's preferences.
VISITOR_INFO1_LIVE
This cookie is set by YouTube. Used to track the information of the embedded YouTube videos on a website.


 LIN ZHENG is a Professor at
LIN ZHENG is a Professor at  PEI HAN is a current PhD student in analytical chemistry at King’s College London, under the supervision of Dr Cristina Legido-Quigley. She studied traditional Chinese medicine for her first degree and now focuses on the application of metabolomics in Chinese herbal medicine research.
PEI HAN is a current PhD student in analytical chemistry at King’s College London, under the supervision of Dr Cristina Legido-Quigley. She studied traditional Chinese medicine for her first degree and now focuses on the application of metabolomics in Chinese herbal medicine research. MIN KIM is a final year PhD student at King’s College London. His research/ PhD is on the identification of Alzheimer’s disease biomarkers using metabolomics. To date, he has analysed over 1,500 human blood samples using LC-MS, which has resulted in six publications of which he is the first co-author of two.
MIN KIM is a final year PhD student at King’s College London. His research/ PhD is on the identification of Alzheimer’s disease biomarkers using metabolomics. To date, he has analysed over 1,500 human blood samples using LC-MS, which has resulted in six publications of which he is the first co-author of two.