Protein PEGylation: An overview of chemistry and process considerations
Posted: 22 February 2010 | Professor Conan Fee (University of Canterbury NZ), Vinod Babu Damodaran (University of Canterbury NZ)
Innovative drug delivery technologies are key components of drug development, with commercial and intellectual values. PEGylation is an excellent example of a delivery system that has scientific and multi-billion dollar commercial importance due to the remarkable improvement in the circulatory half lives of therapeutics, especially for proteins and peptides but even for small molecule pharmaceuticals. Beginning with a brief introduction to the pharmaceutical advantages of PEGylated therapeutics, the authors review the development of this technology over the past four decades in terms of conjugation chemistry, poly(ethylene glycol) structure and process considerations, and conclude that improved, versatile and generic production methods are required to meet the growing demands of the pharmaceutical market.
Bioconjugation and polymer therapeutics
The rapid growth in biotechnology and molecular biology during the last two decades has contributed to a substantial increase in the number of biological products such as proteins, peptides, hormones and enzymes for pharmaceutical applications1,2. The pharmaceutical significance of these biomolecules can be mainly attributed to their high specificity, rapid onset of action and requirement for relatively small doses compared to conventional synthetic molecules3. Unfortunately, most biomolecules are characterised by short circulating lives, low stability due to proteolytic and enzymatic degradation in vivo and rapid clearance from the body via glomerular filtration4,5.
To overcome these drawbacks, biomolecules can be protected through covalent binding with another molecule, or bioconjugation6. Many polymers, from both biological and synthetic origins, are used to protect biomolecules. The resulting polymer bioconjugates are characterised by improved properties such as reduced immunogenicity, decreased antibody recognition, increased in vivo residence time, increased drug targeting specificity and bioavailability, and improved pharmacokinetics6-10. The concept of polymer bioconjugation has been extended to small, high-value prodrugs, leading to a new era of polymeric drug delivery systems and polymer therapeutics8. Commonly used polymers in drug delivery applications include poly(N-(2-hydroxypropyl) methacrylamide) (PHPMA), poly(oligoethylene glycol methyl ether methacrylate) (POEGMA), poly(D,L-lactic-co-glycolic acid) (PLGA), poly(glutamic acid) (PGA), poly(N-isopropyl acrylamide) (PNIPAM), poly(N,N’- diethyl acrylamide) (PDEAM), polystyrene and poly(ethylene glycol) (PEG).
PEG – the polymer of choice
PEG is biocompatible, lacks immunogenicity, antigenicity and toxicity, is soluble in water and other organic solvents, is readily cleared from the body and has high mobility in solution, making this the polymer of choice for bioconjugation11-13. The conjugation of a biomolecule with PEG will result in the modification of its physiochemical properties, particularly size, and increase the systemic retention of the therapeutic agent in the body. It may also enable the moiety to cross the cell membrane by endocytosis to reach particular intracellular targets8. Moreover, PEG is one of a small number of synthetic polymers generally regarded as safe by the US FDA for internal administration14.
Protein PEGylation and its pharmaceutical significance
In the late 1970s, Professor Frank Davis and his colleagues covalently linked methoxy PEG (mPEG) to bovine serum albumin15 and bovine liver catalase16, using cyanuric chloride as an activating agent. Their studies showed that “hanging a bit of PEG onto a protein” markedly improved the overall properties and stability of the protein17,18. This technique is now well established and is known as “PEGylation”. The applications of PEGylation can be extended to peptides, enzymes, antibody fragments, nucleotides and even small organic molecules19,20.
PEGylation can impart several significant and distinct pharmacological advantages over the unmodified form, including improved drug solubility, reduced dosage frequency, toxicity and rate of kidney clearance, an extended circulating life, increased drug stability, enhanced protection from proteolytic degradation, decreased immunogenicity and antigenicity, and minimal loss of biological activity4,21-23.
The reduced kidney clearance of PEGylated proteins can be attributed to an apparent shielding of protein surface charges and an increased hydrodynamic volume of the conjugated product24 due to the ability of PEG molecules to coordinate with two to three water molecules per monomer unit25. Furthermore, these hydrated polymer chains provide a protecting mask for the protein, becoming more effective with an increase in the number and molecular weights of the attached PEGs, reducing the phagocytic uptake by parenchimal cells, preventing opsonisation and increasing residence time in systemic circulation24,26.
In addition to these pharmacological advantages, PEGylation can substantially alter the physicochemical properties of the parent protein, including electrostatic and hydrophobic properties23. PEGylation significantly influences the elimination pathway of the molecule, by shifting from a renal to a hepatic pathway. The tissue-organ distribution profile of the molecule is also greatly influenced by PEGylation, wherein PEGylated proteins preferably follow a peripheral distribution24,26. The pharmaceutical value of PEGylation is now well accepted, with many FDA approved drugs already launched in the market and many in clinical trials. Table 1 shows some examples of approved PEGylated therapeutics.
Table 1 – Examples of FDA approved PEGylated drugs
Name | Company | Year | Indication | Therapeutic area |
Cimzia® | Nektar/UCB | 2008 | Crohn’s disease | Immunology |
Mircera® | Nektar/Roche | 2007 | Anemia in chronic kidney disease (CKD) | Nephrology |
Macugen® | OSI (previously EyeTech) | 2004 | Age-related macular degeneration (AMD) | Ocular |
Somavert® | Pfizer | 2003 | Acromegaly | Immunology |
Pegasys® | Roche | 2002 | Hepatitis C | Immunology |
Neulasta® | Amgen | 2002 | Neutropenia during chemotherapy | Oncology |
PEG-Intron® | Schering-Plough/Enzon | 2001 | Hepatitis C | Immunology |
Oncaspar® | Enzon | 1994 | Lymphoblastic leukemia | Oncology |
Adagen® | Enzon | 1990 | Severe combined immunodeficiency disease (SCID) | Oncology |
The chemistry of protein conjugation
PEGylation conjugation strategy has developed from non-specific random conjugations, known as “first generation PEGylation,” to the more recent site-specific conjugation methods known as “second generation PEGylation”27, although both methods continue to be used. The increase in PEGylation specificity can be mainly credited to the availability of more specific functionalisation of PEG molecules capable of reacting to particular functional moieties in the protein. The result is controlled, well-defined conjugated products with improved product profiles over those obtained through non-specific random conjugations.
Amine conjugation
Because of the availability of a number of accessible primary amino groups on the surface of a protein, conjugation through this functional group is the most extensively used method. Lysine, ornithine and N-terminal amino groups are the most commonly exploited28. Early developments in PEGylation mainly targeted the N-terminal amino groups of lysine. The first reactions reported by Davis and his colleagues involved the reaction of cyanuric chloride activated PEG with the primary amine groups of bovine serum albumin15 and bovine liver catalase16, through alkylation of their respective amine terminals (see Figure 1a). Thereafter, PEG-tresylate was also developed for protein conjugation through alkylation29 but all these reactions resulted in non-specific, multiply conjugated products.

Figure 1 – PEGylation process using amine conjugation chemistries
A greater specificity and selectivity in N-alkyl conjugation strategy was developed after the introduction of PEG aldehyde derivatives, particularly mPEG-propionaldehyde30, capable of forming a stable secondary amine linkage with amino groups through reductive alkylation using sodium cyanoborohydride (see Figure 1b). Because the reactivity of aldehyde groups depends on the nucleophilicity of amine groups, reaction will take place only when the pH of the medium is near or above the pKa of that particular amine terminal. Hence, by controlling the pH of the reaction medium, the heterogeneity of the product profile can be greatly reduced12. This conjugation strategy was adopted for the development of Neulasta®, a PEGylated granulocyte colony stimulating factor (PEG-G-CSF), where a linear 20 kDa mPEG-aldehyde derivative was selectively attached to an N-terminal methionine residue of filgrastim through reductive alkylation under mild acidic conditions31,32. Another example using reductive alkylation chemistry is the PEGylation of a recombinant soluble tumour necrosis factor receptor type I (sTNF-RI) from E. coli, for use in treating chronic inflammatory diseases33.
The acylation of the N-terminal amino acids results in the formation of stable amide and urethane linkages. PEG derivatives activated with succinimidyl succinate (PEG-SS)34, succinimidyl carbonate (PEG-SC)35, benzotriazole carbonate (PEG-BTC)36, phenyl carbonate37,38, carbonylimidazole39, and thiazolidine-2-thione40 were used early in protein conjugation, following the N-terminal acylation pathway. Adagen® (PEG-adenosine deaminase) and Oncaspar® (PEG-asparaginase), the first FDA-approved PEGylated products launched in the market, are prepared based on this chemistry41 (see Figure 1c, page 19). PEG-Intron® (PEG-α-interferon 2b) is prepared by conjugating interferon with a single chain 12 kDa PEG-SC via a urethane bond (see Figure 1, page 19), where the PEG is mainly conjugated with the histidine residue42.
The introduction of monosubstituted propionic and butanoic acid PEG derivatives by Harris and Kozlowski43 and their subsequent activation using succinimide derivatives contributed a significant improvement in amine conjugatio. Pegasys®, another PEGylated interferon, is prepared by mono-PEGylation of interferon-α-2a with an N-hydroxysuccinimide (NHS) activated 40 kDa branched PEG molecule44 (see Figure 2). Somavert®, is also prepared by conjugating 4 – 5 NHS activated 5 kDa PEG derivatives with the lysines of human growth hormone antagonist45,46. Due to the selectivity of NHS active esters towards primary amine terminals, this conjugation technique remains one of the most common conjugation strategies47. Even though amine conjugation is widely accepted and clinically proven, the method is associated with some potential disadvantages, including loss of bioactivity due to the inactivation of critical functional groups and the formation of multiply PEGylated products due to the presence of multiple amino groups available for conjugation48.

Figure 2 – Conjunation chemistry for Pegasys 44
Thiol conjugation
Selective thiol conjugation with natural or genetically engineered, unpaired cysteine residues provides a site-specific conjugation methodology. Thiol selective derivatives such as PEG-maleimide, vinylsulfone, iodoacetamide, and orthopyridyl disulfide are used for cysteine conjugation through formation of thioether or disulfide linkages12. Examples using PEG-maleimide include those at the genetically introduced cysteine residue of trichosanthin (TCS) using 5 and 20 kDa49, antitumor necrosis factor-α-scFv fragment (anti-TNF-α-scFv) using 5, 20 and 40 kDa50 and recombinant staphylokinase (Sak) using 5, 10 and 20 kDa derivatives51 (see Figure 3).

Figure 3 – Cysteine conjugation using PEG-maleimide
Because of the limited availability of single cysteine residues and the chances of protein dimerisation resulting from the introduction of genetically engineered cysteines, the use of this strategy has been limited. Taking advantage of a higher number of accessible disulfide linkages present with paired cysteines in proteins, Balan et al. reported a thiol specific bis-alkylation PEGylation, with the two sulphur atoms which were generated by the mild reduction of these disulfide bonds52,53. A selective reduction of the disulfide bridges was performed using dithiothreitol (DTT) in neutral conditions or tris-(2-carboxyethyl) phosphine hydrochloride (TCEP-HCl) in slightly acidic conditions. The reduced protein was then treated with an active ester PEG-monosulfone at an acidic pH (see Figure 4, page 20). Steric shielding by the attached PEG molecule against a second molecule approaching the same reduced site, an important requirement for the bridged structure, prevented heterogeneity of the product profile and resulted predominantly in the mono-PEGylated derivative. Retention of the protein’s tertiary structure and selectivity and reversibility of the thiol conjugation are some of the attractive features of this technique.
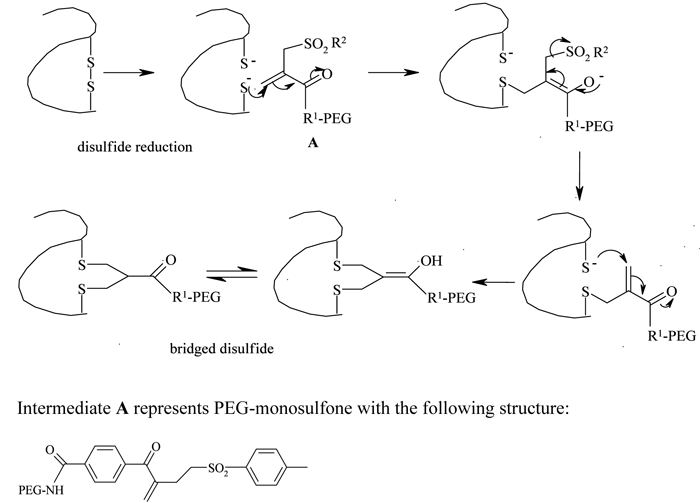
Figure 4 – Disulfide bridging PEGylation and structure of PEG-monosulfone
Oxidised carbohydrate or N-terminal conjugation
The enzymatic (e.g., glucose oxidase) or chemical (e.g., sodium periodate) oxidation of carbohydrate groups present in glycoproteins or N-terminal serine or threonine residues generates reactive aldehyde groups, which can be further conjugated with PEG hydrazide or amine derivatives12,47,54 (see Figure 5).

Figure 5 – Oxidised carbohydrate or N-terminal conjugation
Zalipsky and his colleagues used this methodology for PEGylating immunoglobulin G (IgG), which contains nearly 4% carbohydrate. IgG was first oxidised with periodate and then conjugated with mPEG-hydrazide derivative54. Following the same chemistry, periodate oxidised Ricin A-chain (RTA), an excellent immunotoxin source, was successfully conjugated with a 5 kDa mPEG-hydrazide55.
Transglutaminase (TGase) mediated enzymatic conjugation
A novel, site specific PEGylation methodology targeting glutamine residues was reported by Sato56, using a TGase catalysed acyl transfer reaction between the glutamine (Gln) terminal and PEG primary amino group. Sato developed this distinctive strategy by incorporating a short sequence of Gln residues at the protein terminal, without disturbing its flexibility and conformation, and modified it with primary amine derivatives of PEGs in the presence of TGase (see Figure 6). Compared with other methodologies, TGase mediated conjugations were found to be more site-specific, reliable, reproducible and versatile57. TGase catalysed selective PEGylations of apomyoglobin (apoMb), α-lactalbumin (α-LA), human growth hormone (hGH) human granulocyte colony-stimulating factor (hG-CSF) and human interlukin-2 (hIL-2) with PEG amines are some of the proven examples of this new technique57,58.

Figure 6 – TGase catalysed conjugation
Miscellaneous conjugation chemistries
DeFrees et al59 reported a new site specific process known as GlycoPEGylation, using an enzymatic N-acetylgalactosamine (GalNAc) O-glycolization, followed by PEGylation of the introduced O-glycans using a PEG sailic acid derivative. The new click chemistry strategies60 may also play an emerging role in PEGylation61. Deiters et al62 reported site specific mono-PEGylation of genetically modified superoxide dismutase (SOD) using a PEG-alkyne derivative to attach to the azide terminal (see Figure 7, page 23). Although it has attracted little attention and has limited direct pharmaceutical relevance, PEGylation of human serum albumin using PEG-phenyl-isothiocyanate63 and PEG-epoxide64 may be of scientific interest in developing PEGylation technologies.

Figure 7 – Site specific PEGylation using click chemistry
Reversible PEGylation
The improved physicochemical properties of protein PEGylation are offset in many cases by a substantial reduction in the in vitro protein activity arising from the permanent linkages formed during PEG conjugation. Consequently, a reversible (or releasable) PEGylation concept has been formulated, in which proteins are attached to PEG derivatives through cleavable linkages, which release the protein in vivo at a predetermined kinetic rate65. One example is the release of PEGylated lysozyme using a 1,6-benzyl elimination mechanism66,67 (see Figure 8). A number of similar reversible PEG derivatives have been reported, using bicin, oligo-lactic acid ester, succinic ester, disulfide and β-alanine ester linkers68,69. However, even though this technique potentially provides controlled release of proteins in their fully active forms, the chances of undesirable residual tags remaining with the protein and/or the in vivo formation of reaction by-products remain areas of great concern with this technique.

Figure 8 – Reversible PEGylation using 1,6-benzyl elimination
Structure of PEGs
The development of PEGylation is also characterised by a marked improvement and diversity in the nature of the PEGs used for protein conjugation. Early stage PEGylations were performed mainly with linear and low molecular weight PEGs. The earliest PEG derivatives were also characterised by higher diol contents (around 15%) and polydispersity, contributing to an increased product heterogeneity12,27. The recent commercial availability of pure and low polydisperse PEG derivatives has resulted in much improved product profiles.
The introduction of branched PEG derivatives resulted in an improvement in pharmacological properties, the reasons for which are not yet clear. It has been shown that the net size of linear- and branched-PEG protein conjugates are closely similar70, if not identical, so it is likely that more effective local surface masking and slight decreases in PEG chain flexibility better protect the conjugated protein from proteolytic attack, rather than alter glomerular filtration. The first branched PEG derivative for protein conjugation was prepared by Matsushima et al.71, using the reaction between cyanuric chloride and mPEG, and the resulting species were conjugated with E. coli asparaginase (see Figure 9). Another branched PEG derivative, mPEG2-COOSu, prepared by linking mPEG to both α and ε amino groups of lysine72, forms the basic conjugation strategy for manufacturing Pegasys® (see Figure 2, page 20).

Figure 9 – PEGylation using cyanuric chloride activated branched PEG
The introduction of PEG dendrimers resulted in increased protection due to the addition of very high molecular weights. In one such instance, Meireles et al73 introduced a branched PEG derivative having four mPEG branches, with a terminal COOH group available for protein conjugation (see Figure 10, page 24).
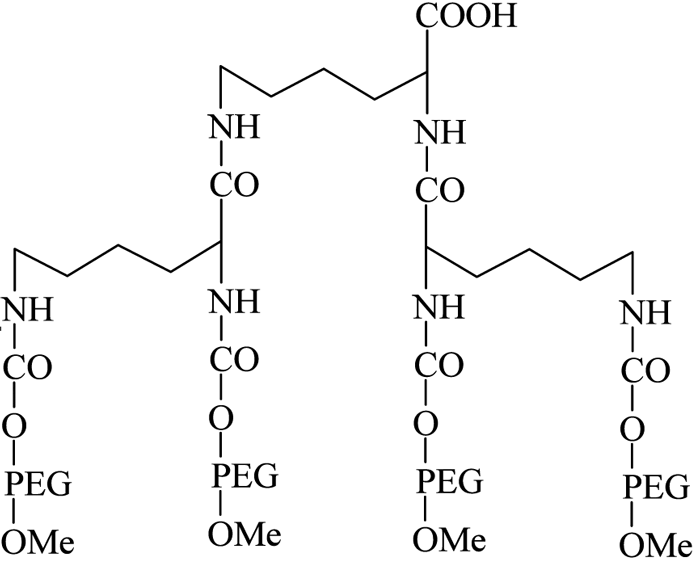
Figure 10 – Four-branched mPEG-COOH dendrimer derivative
They successfully conjugated this with a number of therapeutic proteins, including IFN-α2b, recombinant streptokinase (r-SK), erythropoietin (EPO), granulocyte-colony stimulating factor (G-CSF) and epidermal growth factor (EGF), through NHS activation and found improved pharmacological properties for these products compared with those obtained from two branched-structure of similar molecular mass. Recent developments in the synthesis of linear and branched multifunctional PEG derivatives, known as MultiPEGs (see Figure 11, page 24), have contributed a substantial improvement in the pharmacological advantages of PEGylation technology74-78. However, one disadvantage of very high molecular weight PEGylation is that large, inert PEG molecules are not easily cleared by glomerular filtration and therefore remain in circulation long after the protein moiety has been eliminated through proteolysis. Thus, the effects of long-term PEG retention in vivo should be carefully considered, particularly for therapeutics that require prolonged treatment regimens.
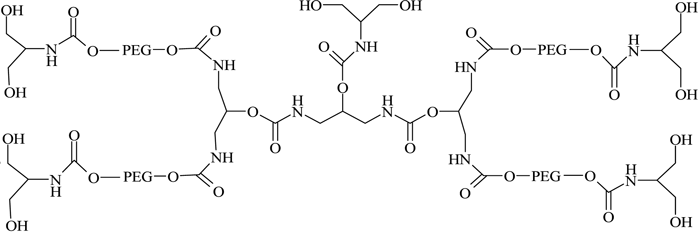
Figure 11 – A MultiPEG derivative (TetraPEG-(OH)10)75
Process considerations
The overall PEGylation processes used to date for protein conjugation can be broadly classified into two types, namely a solution phase batch process and an on-column fed-batch process79. The simple and commonly adopted batch process involves the mixing of reagents together in a suitable buffer solution, preferably at a temperature between 4 and 6°C, followed by the separation and purification of the desired product using a suitable technique based on its physicochemical properties, including sixe exclusion chromatography (SEC), ion exchange chromatography (IEX), hydrophobic interaction chromatography (HIC), membranes or aqueous two phase systems80. A detailed discussion of the separation and purification of PEGylated products is beyond the scope of this paper and we restrict our discussion below to techniques that have been used to affect conjugation.
Normally with the batch process, prolonged contact between reacting species and products results in multiple conjugations and gives rise to a number of PEG isomers. Consequently a heterogeneous product mixture results, constituting unreacted starting materials, hydrolysed activating agents and a wide range of PEGylated products with varying degrees of conjugation. A typical SEC elution profile for various PEGylated α-lactalbumin products obtained from a batch process is illustrated in Figure 12 (see page 24. Hence, extensive multistep purifications and downstream processing are required to isolate the desired product79, significantly decreasing overall yields. The high cost of the therapeutic proteins, along with the cost of separating the desired PEGylated protein from the reaction mixtures, makes the products extremely expensive.
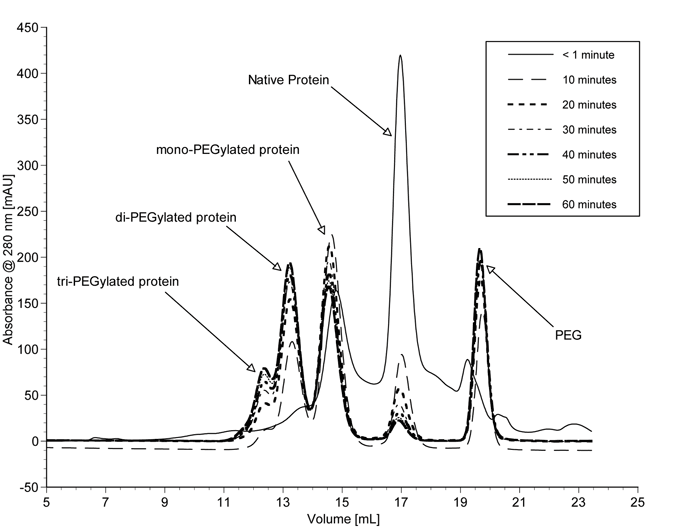
Figure 12 – SEC elution profile of various PEGylated a-lactalbumin products obtained from batch process 87
A number of on-column PEGylation techniques have been attempted recently, with an emphasis on improving the product profile and specificity of conjugation. The first such approach was reported by Felix et al81,82, who developed a site-specific solid phase peptide PEGylation, in which the peptide sequence was tethered onto a Rink amide MBHA-resin and was conjugated with a PEG derivative through a side chain lysine or aspartic acid. Finally, the mono-PEGylated peptide was cleaved off from the resin using trifluoroacetic acid (TFA). In a similar way, folate was derivatised with a PEG derivative attached to HMP resin through a peptide linker and the peptide-PEG-folate conjugate was cleaved off the resin using TFA83. Solid-phase synthesis is not practical for large polypeptides (proteins) and the harsh chemicals, such as TFA, required for the release of solid-linked PEGylated products means that direct application of this methodology is not viable with highly sensitive species.
Utilising the ion exchange interactions between protein and ion exchange resins, Monkarsh et al84 prepared and separated various positional isomers of PEGylated interferone-α-2a (PEG-INF) using an on-column process. INF was first adsorbed to a strong anion exchange resin and the activated PEG derivative was then circulated through the column. After eluting the unreacted PEGs and by-products, PEG-INF was collected by increasing the salt concentration. Unfortunately, this method resulted in multi-PEGylated products, without any improvement in the product profile.
Similarly, Lee et al85 PEGylated recombinant interferon-α-2a (rhIFN-α-2a) by adsorbing on a CM-Sepharose cation exchange resin through ion exchange interactions. After washing out the unbound rhIFN-α-2a, mPEG-aldehyde and sodium cyanoborohydride solutions were passed through the column, resulting in N-terminal PEGylation. Suo et al86 PEGylated bovine haemoglobin loaded onto CM-Sepharose using mPEG-succinimidyl carbonate and their studies showed that the selectivity of this method was increased by an increase in the molecular weight of the conjugated PEG. Even though these methods resulted in mono-PEGylated products at up to 75% yield, based on the loaded native protein, consistency in the reaction efficiency remains a challenge, due to the random orientations of adsorbed protein on the ion exchange matrix.
A unique on-column PEGylation methodology, known as size exclusion reaction chromatography (SERC), was introduced by Fee87, incorporating the principle of SEC in separating various molecular sized species based on their different linear velocities through a column packed with porous beads. In this method, activated PEG and protein form a transient in-situ moving reaction zone within the column, in which the PEGylated protein, having a larger size than either of the reagents, moves ahead of the reaction zone, thus limiting its residence time in contact with activated PEG and reducing over-PEGylation. This process, however, is unable to control positional isomers.
Finally, some solution-phase88 and on-column89 attempts have been made to protect the active site of a protein by conjugating in the presence of an affinity partner. In this approach, which appears to be effective in some cases, the inherent steric hindrance of the protein complexed with its binding partner is used to preferentially PEGylate areas away from the active site and thus retain the in vitro activity of the released PEG-protein.
Conclusion
Nearly four decades of development in PEGylation technology has proven its pharmacological advantages and acceptability but the technology still lags in providing a commercially attractive, generic process to produce highly specific PEGylated therapeutic products at high yield. As a multi-million dollar annual business with the growing interest from both emerging biotechnology and established multinational pharmaceutical companies, there is great scientific and commercial interest in improving present methodologies and in introducing innovative process variations.
References
- Lubiniecki, A. S., Potential influence of international harmonization of pharmaceutical regulations on biopharmaceutical development. Current Opinion in Biotechnology 1997, 8, (3), 350-356.
- Blohm, D.; Bollschweiler, C.; Hillen, H., Pharmaceutical Proteins. Angewandte Chemie International Edition in English 1988, 27, (2), 207-225.
- Wu-Pong, S.; Rojanasakul, Y., Biopharmaceutical Drug Design and Development. Humana, Totowa, N. J.: 1999; 435 pp.
- Nucci, M. L.; Shorr, R.; Abuchowski, A., The therapeutic value of poly(ethylene glycol)-modified proteins. Advanced Drug Delivery Reviews 1991, 6, (2), 133-151.
- Pasut, G.; Veronese, F. M., Polymer-drug conjugation, recent achievements and general strategies. Progress in Polymer Science 2007, 32, (8-9), 933-961.
- Veronese, F. M.; Morpurgo, M., Bioconjugation in pharmaceutical chemistry. Il Farmaco 1999, 54, (8), 497-516.
- Lutz, J.-F.; Borner, H. G., Modern trends in polymer bioconjugates design. Progress in Polymer Science 2008, 33, (1), 1-39.
- Khandare, J.; Minko, T., Polymer-drug conjugates: Progress in polymeric prodrugs. Progress in Polymer Science 2006, 31, (4), 359-397.
- Duncan, R., The dawning era of polymer therapeutics. Nature Reviews. Drug Discovery 2003, 2, (5), 347.
- Gaspar, R.; Duncan, R., Polymeric carriers: Preclinical safety and the regulatory implications for design and development of polymer therapeutics. Advanced Drug Delivery Reviews 2009, 61, (13), 1220-1231.
- Pasut, G.; Veronese, M. F., PEGylation of Proteins as Tailored Chemistry for Optimized Bioconjugation. Advances in Polymer Sciences (Polymer Theraputics I) 2006, 192, 95-134.
- Roberts, M. J.; Bentley, M. D.; Harris, J. M., Chemistry for peptide and protein PEGylation. Advanced Drug Delivery Reviews 2002, 54, (4), 459-476.
- Harris, J. M., Introduction to biotechnical and biomedical applications of poly(ethylene glycol). Poly(Ethylene Glycol) Chemistry Biotechnical and biomedical applications 1992, 1-14.
- Bhattarai, N.; Matsen, F. A.; Zhang, M., PEG-Grafted Chitosan as an Injectable Thermoreversible Hydrogel. Macromolecular Bioscience 2005, 5, (2), 107-111.
- Abuchowski, A.; Van Es, T.; Palczuk, N. C.; Davis, F. F., Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. The Journal of Biological Chemistry 1977, 252, (11), 3578-3581.
- Abuchowski, A.; McCoy, J. R.; Palczuk, N. C.; Van Es, T.; Davis, F. F., Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. The Journal of Biological Chemistry 1977, 252, (11), 3582-3586.
- Katsnelson, A., Profile: Abe Abuchowski. Nature Biotechnology 2006, 24, (9), 1050-1050.
- Davis, F. F., The origin of pegnology. Advanced Drug Delivery Reviews 2002, 54, (4), 457-458.
- Riley, T.; Riggs-Sauthier, J., The Benefits and Challenges of PEGylating Small Molecules. Pharmaceutical Technology 2008, 32, (7), 88-94.
- Matsushima, A.; Kodera, Y.; Hiroto, M.; Nishimura, H.; Inada, Y., Bioconjugates of proteins and polyethylene glycol: potent tools in biotechnological processes. Journal of Molecular Catalysis B: Enzymatic 1996, 2, (1), 1-17.
- Veronese, F. M.; Pasut, G., PEGylation, successful approach to drug delivery. Drug Discovery Today 2005, 10, (21), 1451-1458.
- Kozlowski, A.; Harris, J. M., Improvements in protein PEGylation: pegylated interferons for treatment of hepatitis C. Journal of Controlled Release 2001, 72, (1-3), 217-224.
- Bailon, P.; Berthold, W., Polyethylene glycol-conjugated pharmaceutical proteins. Pharmaceutical Science & Technology Today 1998, 1, (8), 352-356.
- Caliceti, P.; Veronese, F. M., Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates. Advanced Drug Delivery Reviews 2003, 55, (10), 1261-1277.
- Antonsen, K. P.; Hoffman, A. S., Water structure of PEG solutions by differential scanning calorimetry measurements. Plenum Press, New York: 1992; 15-28.
- Hamidi, M.; Azadi, A.; Rafiei, P., Pharmacokinetic Consequences of Pegylation. Drug Delivery 2006, 13, (6), 399-409.
- Pasut, G.; Veronese, F. M., PEGylation of proteins as tailored chemistry for optimized bioconjugates. Advances in Polymer Science 2006, 192, (Polymer Therapeutics I), 95-134.
- Bruckdorfer, T., Drug delivery with PEGylaton. European Biopharmaceutical Review 2008, (Spring), 96-104.
- Gais, H.-J.; Ruppert, S., Modification and immobilization of proteins with polyethylene glycol tresylates and polysaccharide tresylates: evidence suggesting a revision of the coupling mechanism and the structure of the polymer-polymer linkage. Tetrahedron Letters 1995, 36, (22), 3837-8.
- Harris, J. M.; Sedaghat-Herati, M. R. Preparation and use of polyethylene glycol propionaldehyde for modification of proteins. US5252714, 1993.
- Veronese, F. M.; Mero, A., The impact of PEGylation on biological therapies. BioDrugs 2008, 22, (5), 315-329.
- Kinstler, O.; Molineux, G.; Treuheit, M.; Ladd, D.; Gegg, C., Mono-N-terminal poly(ethylene glycol)-protein conjugates. Advanced Drug Delivery Reviews 2002, 54, (4), 477-485.
- Edwards, C. K.; Martin, S. W.; Seely, J.; Kinstler, O.; Buckel, S.; Bendele, A. M.; Ellen Cosenza, M.; Feige, U.; Kohno, T., Design of PEGylated soluble tumor necrosis factor receptor type I (PEG sTNF-RI) for chronic inflammatory diseases. Advanced Drug Delivery Reviews 2003, 55, (10), 1315-1336.
- Zalipsky, S.; Lee, C., Use of functionalized poly(ethylene glycol)s for modification of polypeptides. In Poly(ethylene glycol) chemistry Biotechnical and Biomedical applications, Harris, J. M., Ed. Plenum Press, NY: 1992; 347-70.
- Zalipsky, S.; Seltzer, R.; Nho, K., Succinimidyl carbonates of polyethylene glycol. Useful reactive polymers for preparation of protein conjugates. ACS Symposium Series 1991, 469, (Polymeric Drugs and Drug Delivery Systems), 91-100.
- Dolence, E. K.; Hu, C.-Z.; Tsang, R.; Sanders, C. G.; Osaki, S. Electrophilic polyethylene oxides for the modification of polysaccharides, polypeptides (proteins) and polymer surfaces. WO9607670, 1996.
- Woodward, C. A.; Kaufman, E. N., Enzymic catalysis in organic solvents: polyethylene glycol modified hydrogenase retains sulfhydrogenase activity in toluene. Biotechnology and Bioengineering 1996, 52, (3), 423-428.
- Veronese, F. M.; Largajolli, R.; Boccu, E.; Benassi, C. A.; Schiavon, O., Surface modification of proteins. Activation of monomethoxy-polyethylene glycols by phenylchloroformates and modification of ribonuclease and superoxide dismutase. Applied Biochemistry and Biotechnology 1985, 11, (2), 141-52.
- Beauchamp, C. O.; Gonias, S. L.; Menapace, D. P.; Pizzo, S. V., A new procedure for the synthesis of polyethylene glycol-protein adducts; effects on function, receptor recognition, and clearance of superoxide dismutase, lactoferrin, and alpha 2-macroglobulin. Analytical Biochemistry 1983, 131, (1), 25-33.
- Greenwald, R. B.; Pendri, A.; Martinez, A.; Gilbert, C.; Bradley, P., PEG Thiazolidine-2-thione, a Novel Reagent for Facile Protein Modification: Conjugation of Bovine Hemoglobin. Bioconjugate Chemistry 1996, 7, (6), 638-641.
- Davis, F. F., PEG-adenosine deaminase and PEG-asparaginase. Advances in Experimental Medicine and Biology 2003, 519, (Polymer Drugs in the Clinical Stage), 51-58.
- Wang, Y.-S.; Youngster, S.; Grace, M.; Bausch, J.; Bordens, R.; Wyss, D. F., Structural and biological characterization of pegylated recombinant interferon alpha-2b and its therapeutic implications. Advanced Drug Delivery Reviews 2002, 54, (4), 547-570.
- Harris, J. M.; Kozlowski, A. Preparation of poly(ethylene glycol) monosubstituted with propionic or butanoic acids and functional derivatives thereof for biotechnical applications. US5672662, 1997.
- Bailon, P.; Palleroni, A.; Schaffer, C. A.; Spence, C. L.; Fung, W.-J.; Porter, J. E.; Ehrlich, G. K.; Pan, W.; Xu, Z.-X.; Modi, M. W.; Farid, A.; Berthold, W.; Graves, M., Rational Design of a Potent, Long-Lasting Form of Interferon: A 40 kDa Branched Polyethylene Glycol-Conjugated Interferon α-2a for the Treatment of Hepatitis C. Bioconjugate Chemistry 2001, 12, (2), 195-202.
- Olson, K.; Gehant, R.; Mukku, V.; O’Connell, K.; Tomlinson, B.; Totpal, K.; Winkler, M., Preparation and characterization of poly(ethylene glycol)ylated human growth hormone antagonist. ACS Symposium Series 1997, 680,
- Parkinson, C.; Scarlett, J. A.; Trainer, P. J., Pegvisomant in the treatment of acromegaly. Advanced Drug Delivery Reviews 2003, 55, (10), 1303-1314.
- Hermanson, G. T., Bioconjugate Techniques. Academic Press, San Diego: 1996; 786 pp.
- Zappe, H.; Snell, M. E.; Bossard, M. J., PEGylation of cyanovirin-N, an entry inhibitor of HIV. Advanced Drug Delivery Reviews 2008, 60, (1), 79-87.
- He, X.-H.; Shaw, P.-C.; Tam, S.-C., Reducing the immunogenicity and improving the in vivo activity of trichosanthin by site-directed PEGylation. Life Sciences 1999, 65, (4), 355-368.
- Yang, K.; Basu, A.; Wang, M.; Chintala, R.; Hsieh, M.-C.; Liu, S.; Hua, J.; Zhang, Z.; Zhou, J.; Li, M.; Phyu, H.; Petti, G.; Mendez, M.; Janjua, H.; Peng, P.; Longley, C.; Borowski, V.; Mehlig, M.; Filpula, D., Tailoring structure-function and pharmacokinetic properties of single-chain Fv proteins by site-specific PEGylation. Protein Engineering 2003, 16, (10), 761-770.
- Moreadith, R. W.; Collen, D., Clinical development of PEGylated recombinant staphylokinase (PEG-Sak) for bolus thrombolytic treatment of patients with acute myocardial infarction. Advanced Drug Delivery Reviews 2003, 55, (10), 1337-1345.
- Balan, S.; Choi, J.; Godwin, A.; Teo, I.; Laborde, C. M.; Heidelberger, S.; Zloh, M.; Shaunak, S.; Brocchini, S., Site-Specific PEGylation of Protein Disulfide Bonds Using a Three-Carbon Bridge. Bioconjugate Chemistry 2007, 18, (1), 61-76
- Brocchini, S.; Godwin, A.; Balan, S.; Choi, J.-w.; Zloh, M.; Shaunak, S., Disulfide bridge based PEGylation of proteins. Advanced Drug Delivery Reviews 2008, 60, (1), 3-12.
- Zalipsky, S.; Menon-Rudolph, S., Hydrazide derivatives of poly(ethylene glycol) and their bioconjugates. ACS Symposium Series 1997, 680, (Poly(ethylene glycol) Chemistry and biological applications), 318-341.
- Youn, Y. S.; Na, D. H.; Yoo, S. D.; Song, S.-C.; Lee, K. C., Carbohydrate-specifically polyethylene glycol-modified ricin A-chain with improved therapeutic potential. The International Journal of Biochemistry & Cell Biology 2005, 37, (7), 1525-1533.
- Sato, H., Enzymatic procedure for site-specific pegylation of proteins. Advanced Drug Delivery Reviews 2002, 54, (4), 487-504.
- Fontana, A.; Spolaore, B.; Mero, A.; Veronese, F. M., Site-specific modification and PEGylation of pharmaceutical proteins mediated by transglutaminase. Advanced Drug Delivery Reviews 2008, 60, (1), 13-28.
- Mero, A.; Spolaore, B.; Veronese, F. M.; Fontana, A., Transglutaminase-Mediated PEGylation of Proteins: Direct Identification of the Sites of Protein Modification by Mass Spectrometry using a Novel Monodisperse PEG. Bioconjugate Chemistry 2009, 20, (2), 384-389.
- DeFrees, S.; Wang, Z.-G.; Xing, R.; Scott, A. E.; Wang, J.; Zopf, D.; Gouty, D. L.; Sjoberg, E. R.; Panneerselvam, K.; Brinkman-Van der Linden, E. C. M.; Bayer, R. J.; Tarp, M. A.; Clausen, H., GlycoPEGylation of recombinant therapeutic proteins produced in Escherichia coli. Glycobiology 2006, 16, (9), 833-843.
- Hein, C. D.; Liu, X.-M.; Wang, D., Click chemistry, a powerful tool for pharmaceutical sciences. Pharmaceutical Research 2008, 25, (10), 2216-2230.
- Droumaguet, B. L.; Velonia, K., Click Chemistry: A Powerful Tool to Create Polymer-Based Macromolecular Chimeras. Macromolecular Rapid Communications 2008, 29, (12-13), 1073-1089.
- Deiters, A.; Cropp, T. A.; Summerer, D.; Mukherji, M.; Schultz, P. G., Site-specific PEGylation of proteins containing unnatural amino acids. Bioorganic & Medicinal Chemistry Letters 2004, 14, (23), 5743-5745.
- Meng, F.; Manjula, B. N.; Smith, P. K.; Acharya, S. A., PEGylation of Human Serum Albumin: Reaction of PEG-Phenyl-Isothiocyanate with Protein. Bioconjugate Chemistry 2008, 19, (7), 1352-1360.
- Bergstrom, K.; Holmberg, K., Protein immobilization to polystyrene via long poly(ethylene lycol) chains. Biotechnology and Bioengineering 1991, 38, (8), 952-5.
- Veronese, F. M.; Pasut, G., PEGylation: Posttranslational bioengineering of protein biotherapeutics. Drug Discovery Today: Technologies 2008, 5, (2-3), e57-e64
- Lee, S.; Greenwald, R. B.; McGuire, J.; Yang, K.; Shi, C., Drug Delivery Systems Employing 1,6-Elimination: Releasable Poly(ethylene glycol) Conjugates of Proteins. Bioconjugate Chemistry 2001, 12, (2), 163-169.
- Greenwald, R. B.; Yang, K.; Zhao, H.; Conover, C. D.; Lee, S.; Filpula, D., Controlled Release of Proteins from Their Poly(Ethylene Glycol) Conjugates: Drug Delivery Systems Employing 1,6-Elimination. Bioconjugate Chemistry 2003, 14, (2), 395-403.
- Filpula, D.; Zhao, H., Releasable PEGylation of proteins with customized linkers. Advanced Drug Delivery Reviews 2008, 60, (1), 29-49.
- Pasut, G.; Mero, A.; Caboi, F.; Scaramuzza, S.; Sollai, L.; Veronese, F. M., A New PEG beta -Alanine Active Derivative for Releasable Protein Conjugation. Bioconjugate Chemistry 2008, 19, (12), 2427-2431.
- Fee, C. J., Size comparison between proteins PEGylated with branched and linear poly(ethylene glycol) molecules. Biotechnology and Bioengineering 2007, 98, (4), 725-731.
- Matsushima, A.; Nishimura, H.; Ashihara, Y.; Yokota, Y.; Inada, Y., Modification of E. coli asparaginase with 2,4-bis(O-methoxypolyethylene glycol)-6-chloro-s-triazine (activated PEG2); disappearance of binding ability towards anti-serum and retention of enzymic activity. Chemistry Letters 1980, (7), 773-6.
- Monfardini, C.; Schiavon, O.; Caliceti, P.; Morpurgo, M.; Harris, J. M.; Veronese, F. M., A Branched Monomethoxypoly(ethylene glycol) for Protein Modification. Bioconjugate Chemistry 2002, 6, (1), 62-69.
- Meireles, R. P.; Hernandez, J. A. R.; Odio, F. R. C.; Martinez, V. M. S.; Sanchez, E. F. Four branched dendrimer-PEG for conjugation to proteins and peptides. EP1967212, 2008.
- Zacchigna, M.; Di Luca, G.; Cateni, F.; Maurich, V.; Ballico, M.; Bonora, G. M.; Drioli, S., New MultiPEG-conjugated theophylline derivatives: Synthesis and pharmacological evaluations. European Journal of Pharmaceutical Sciences 2007, 30, (3-4), 343-350.
- Drioli, S.; Bonora, G. M.; Ballico, M., New syntheses of branched, multifunctional high-molecular weight poly(ethylene glycol)s or (MultiPEG)s. Open Organic Chemistry Journal 2008, 2, 17-25.
- Liu, X.-M.; Thakur, A.; Wang, D., Efficient Synthesis of Linear Multifunctional Poly(ethylene glycol) by Copper(I)-Catalyzed Huisgen 1,3-Dipolar Cycloaddition. Biomacromolecules 2007, 8, (9), 2653-2658.
- Pasut, G.; Scaramuzza, S.; Schiavon, O.; Mendichi, R.; Veronese, F. M., PEG-epirubicin conjugates with high drug loading. Journal of Bioactive and Compatible Polymers 2005, 20, (3), 213-230.
- Schiavon, O.; Pasut, G.; Moro, S.; Orsolini, P.; Guiotto, A.; Veronese, F. M., PEG-Ara-C conjugates for controlled release. European Journal of Medicinal Chemistry 2004, 39, (2), 123-133.
- Fee, C. J.; Van Alstine, J. M., PEG-proteins: Reaction engineering and separation issues. Chemical Engineering Science 2006, 61, (3), 924-939.
- Fee, C. J., Protein conjugates purification and characterization. PEGylated Protein Drugs: Basic Science and Clinical Applications, Veronese, F. M., Ed. Birkhauser Publishing: Basel, 2009; 113-125.
- Felix, A. M., Site-specific poly(ethylene glycol)ylation of peptides. ACS Symposium Series 1997, 680, (Poly(ethylene glycol) Chemistry and biological applications), 218-238.
- Lu, Y.-A.; Felix, A. M., Pegylated peptides. III. Solid-phase synthesis with pegylating reagents of varying molecular weight: synthesis of multiply pegylated peptides. Reactive Polymers 1994, 22, (3), 221-9.
- Niculescu-Duvaz, D.; Getaz, J.; Springer, C. J., Long Functionalized Poly(ethylene glycol)s of Defined Molecular Weight: Synthesis and Application in Solid-Phase Synthesis of Conjugates. Bioconjugate Chemistry 2008, 19, (4), 973-981.
- Monkarsh, S. P.; Spence, C.; Porter, J. E.; Palleroni, A.; Nalin, C.; Rosen, P.; Bailon, P., Isolation of positional isomers of monopoly(ethylene glycol)ylated interferon alpha -2a and the determination of their biochemical and biological characteristics. ACS Symposium Series 1997, 680, (Poly(ethylene glycol) Chemistry and biological applications), 207-216.
- Lee, B. K.; Kwon, J. S.; Kim, H. J.; Yamamoto, S.; Lee, E. K., Solid-Phase PEGylation of Recombinant Interferon alpha-2a for Site-Specific Modification: Process Performance, Characterization, and in Vitro Bioactivity. Bioconjugate Chemistry 2007, 18, 1728-1734.
- Suo, X.; Zheng, C.; Yu, P.; Lu, X.; Ma, G.; Su, Z., Solid phase pegylation of hemoglobin. Artificial Cells, Blood Substitutes and Biotechnology 2009, 37, (4), 147-155.
- Fee, C. J., Size-exclusion reaction chromatography (SERC): A new technique for protein PEGylation. Biotechnology and Bioengineering 2003, 82, (2), 200-206.
- Salmaso, S.; Semenzato, A.; Bersania, S.; Chinol, M.; Paganelli, G.; Caliceti, P., Preparation and characterization of active site protected poly(ethylene glycol)-avidin bioconjugates. Biochimica et Biophysica Acta, General Subjects 2005, 1726, (1), 57-66.
- Baran, E. T.; Ozer, N.; Hasirci, V., Solid-phase enzyme modification via affinity chromatography. Journal of Chromatography, B Analytical Technologies in the Biomedical and Life Sciences 2003, 794, (2), 311-322.
About the authors

Vinod Babu Damodaran, University of Canterbury, New Zealand
Vinod Babu Damodaran is a senior PhD student working under Professor Fee on improving the yield and selectivity of protein PEGylation. Before this he was a Senior Research Chemist at the Merck Development Centre, India. His background is organic chemistry and his interests include polymer therapeutics, bioconjugation and controlled drug delivery.

Professor Conan Fee, University of Canterbury, New Zealand
Conan Fee is a Professor of Chemical Engineering and is also Co-Director of the Biomolecular Interactions Centre at the University of Canterbury. He holds a PhD in Chemical Engineering and a Diploma in Strategic Management and Leadership. He has research interests in bioseparations, protein PEGylation, drug delivery and biomolecular interactions and has been a consultant in bioseparation process development for a number of companies internationally.