The Sequencing Revolution: enabling personal genomics and personalised medicine
1
SHARES
Posted: 29 October 2010 |
It has been 10 years since the completion of the first draft of the human genome. Today, we are in the midst of a full assault on the human genetic code, racing to uncover the genetic mechanisms that affect disease, aging, happiness, violence … and just about every imaginable human variation. Advances in DNA sequencing technology have enabled individuals to have their own genomes sequenced rapidly, cheaply and in astonishing detail. The sequencing revolution is also changing the way the pharmaceutical industry develops, tests and targets new medicines.
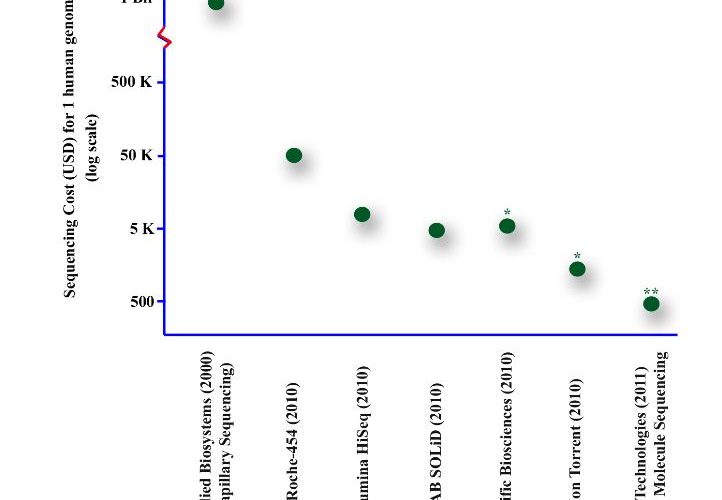
Figure 1 Advances in the development of sequencing technologies have resulted in an increase in data output with a dramatic decrease in cost. This graph compares calculated sequencing costs for one complete haploid human genome sequence (23 chromosomes, three billion bases) * estimated from literature ** marketing figures
It has been 10 years since the completion of the first draft of the human genome. Today, we are in the midst of a full assault on the human genetic code, racing to uncover the genetic mechanisms that affect disease, aging, happiness, violence … and just about every imaginable human variation. Advances in DNA sequencing technology have enabled individuals to have their own genomes sequenced rapidly, cheaply and in astonishing detail. The sequencing revolution is also changing the way the pharmaceutical industry develops, tests and targets new medicines.
This webinar showcases the Growth Direct System; an RMM (Rapid Microbial Method) that improves on traditional membrane filtration, delivering increased accuracy, a faster time to result, enhanced data integrity compliance, and more control over the manufacturing process.
Key learning points:
Understand the benefits of full workflow microbiology quality control testing automation in radiopharmaceutical production
Learn about ITM’s implementation journey and considerations when evaluating the technology
Find out how the advanced optics and microcolony detection capabilities of Growth Direct® technology impact time to result (TTR).
Don’t miss your chance to learn from experts in the industry –Register for FREE
Although humans have recognised hereditary traits in plants, animals and humans for thousands of years, it was only at the beginning of the 20th Century that the concept of the gene arose, when researchers working on fruit flies narrowed down the material basis of trans – missible traits to chromosomes1. Moreover, it was not until 1953, when Watson and Crick determined the double helix structure of DNA, that nucleic acids and their components became the central focus of genetic research2.
Beginning in the 1960s, the first nucleic acid sequencing attempts used chromatography. While this was neither reliable nor robust enough for routine use, it resulted, in 1972, in the publication of the first complete nucleotide sequence of a gene – from the MS2 virus3. Incredibly, it took only four more years to publish the entire 3,600-base sequence of the MS2 genome. Then, in 1977, Frederik Sanger, at the University of Cambridge, published the chain terminator method of DNA sequencing4 which became the method of choice for routine sequencing in academic centres. In the 1980s, the development of dye terminators (nucleotides with unique fluorescent markers) made it possible to automate Sanger’s method, speeding up the process tremendously. By 2000, when the first draft of the human genome (the 24 chromosome haploid sequence), with three billion base pairs, was sequenced, the project had taken only a decade, but had required hundreds of sequencing machines, employed teams of researchers around the globe, and cost three billion dollars. Part of the reason the sequence was completed so quickly was the race between the public consortium (a worldwide network of research centres) and private efforts led by the Celera Corporation to be the first to sequence and profit from the genome’s information5,6.
During the race to complete the human genome, research centres made technical advances on fluorescence based DNA sequencing – such as pyrosequencing, reversible nucleotide terminators and synthesis of ‘DNA colonies’, which allowed for the commercial realisation of Next Generation Sequencing (NGS).
The first NGS system, unveiled in 2004, was the 454 sequencing platform. This is based on a massively parallel pyrosequencing process, which sequences each DNA strand by synthesising a complementary strand7. For each testing cycle, a fluorescently tagged nucleotide (A, T, C or G) is added to the reaction. Each time the tagged nucleotide is incorporated, it generates a light signal of a specific strength, meaning the strength of the overall signal from a sample is proportional to the number of nucleotides incorporated in it. This value is converted into a nucleotide read. The latest 454 platform, the Genome Sequencer FLX, can sequence 500 million bases per 10 hour run at a cost of USD 7,500. At this rate, an entire human genome can be sequenced in one week at a cost of under USD 50,000.
Several other companies have emerged with platforms that can output more sequence data per run with even lower costs – the strongest competitors in the field include Illumina Solexa8, Applied Biosystems (AB) SOLiD9, Helicos Biosciences10, and Complete Genomics11. Where the Sequencer FLX reads strings averaging 660 bases in length, though, these systems are based on short (10 to 150 base) reads – with higher intrinsic error rates requiring repetitive sampling (or coverage) of the template DNA to build the final consensus sequence. The Illumina and Helicos platforms utilise variations on the same theme, but use reversible fluorescence based single nucleotide terminators for their synthesis. AB SOLiD and Complete Genomics use sequencing by ligation strategy with short fluorescently labelled DNA probes. Similar to the 454 platform, billions of parallel sequencing reactions are conducted and the machine run takes approximately one week. However, for these systems, the sequence cost for one complete human genome is under USD 10,000.
Figure 1 Advances in the development of sequencing technologies have resulted in an increase in data output with a dramatic decrease in cost. This graph compares calculated sequencing costs for one complete haploid human genome sequence (23 chromosomes, three billion bases) * estimated from literature ** marketing figures
Illumina’s current HiSeq platform can output 300 billion bases of reads in a single run. This volume of data output makes it possible to sequence several human diploid genomes on one machine with sufficient coverage to guarantee accuracy. Comparably, Applied Biosystems (part of Life Technologies Corporation) has announced the SOLiD 4 system, which produces 100 billion bases of reads with 99.9 per cent accuracy. This is achieved because SOLiD’s probe hybridisation technique sequences each base twice on the template. For both the HiSeq and the SOLiD 4, the cost for one human diploid sequence, with reliable accuracy, is approximately USD 5,000.
The Helicos platform boasts the only commercially available method for single molecule sequencing10. By eliminating steps used in other systems, it significantly reduces the biases and artefacts associated with sample preparation. It also allows the use of almost any nucleic acid as a starting material – including degraded DNA for applications in forensic research. Helicos has also published articles demonstrating direct RNA sequencing on their platform, raising the possibility of assessing the transcriptome in a single cell12.
Complete Genomics Inc. uses rolling circle template amplification, thereby creating a novel nanoball of DNA captured on a solid surface11. Because of the complexity of this system, the company has adopted a model of providing sequencing services rather than selling platforms. Complete Genomics aims to sequence 1000+ human individuals in 2010 and to expand data output by building satellite sequencing facilities.
Following rapid advancements in Second Generation sequencing, the first Third Generation systems will soon be available. Later this year, Pacific Biosciences13 and Life Technologies14,15 are both scheduled to unveil their single molecule sequencing platforms. Current technologies offer up to several hundred base read lengths and shorter processing time using large samples of genetic material, but Third Generation systems will achieve read lengths of greater than 1000 bases from a single DNA molecule, with less sample processing time and lower reagent costs. Long reads can fully span repetitive or structurally varied genomic regions.
Pacific Biosciences makes use of tiny compartments (called Zero Mode Waveguides) where the fluorescence signal of a single nucleotide can be recorded when it is briefly held in place by a DNA polymerase on the template strand (Figure 2). Life Technologies employs a FRET based system – the DNA polymerase is attached to an energy donor bead that can cause light emission from a fluorescently labelled nucleotide only when it is being incorporated in the DNA strand (Figure 2). As with Second Generation machines, the platforms employ optic systems to record emitted light which is converted to a nucleotide read. Both platforms add ‘redundant sequencing’– the DNA strand is sequenced twice to give the sequence 99.9 per cent accuracy. A third platform, Ion Torrent15 (acquired by Life Technologies), does not use optics, but semiconductor chips (Figure 2). The chips are fabricated to detect the hydrogen ion which is normally released when a polymerase incorporates a single nucleotide into the growing DNA strand. The result is a platform that is easier to manufacture and operate with lower costs and shorter run times. Ion Torrent promotes that their system will sell for USD 50,000 and can deliver sequence data, comparable to other platforms, in one hour15.
Figure 2 The third generation sequencing technologies. A) Pacific Biosciences system uses tiny wells (nanometres in diameter) called ‘Zero Mode Wave Guides’. The small volume allows detection of the light emission from a single nucleotide in the vicinity of the DNA polymerase as it is incorporated into the growing DNA strand. B) Life Technologies employs a five nanometre quantum dot nanocrystals (yellow) attached to the polymerase (white). As fluorescent nucleotides are incorporated into the DNA strand, a FRET [Fluorescence Resonance Energy Transfer signal (shown in green) is generated between the nanocrystals and the base, which is recorded by sensitive optics. C) Ion Torrent technology detects the presence of an H+ ion, which is released after a base is normally added to the DNA strand by a DNA polymerase (in green). The reaction occurs in nanolitre wells. The detection system (shown in panel D) employs an ion sensitive layer (green) built on top of a proprietary ion detector (beige) using semiconductor technology.”
Third Generation platforms promise advances in throughput, scalability, speed and cost over the current methods. However, nanopore based technologies, though further from commercial realisation, have the potential for even greater benefits. For instance, the DNA transistor16, currently under development by IBM Research, will offer true single molecule sequencing by decoding the electrical charge of nucleotides of DNA strands as they are threaded through a 1.5 to 3-nanometre-wide pore in a silicon chip. NABsys of Providence, Rhode Island, is testing a system called ‘Hybridisation-assisted Nanopore Sequencing Platform’17 which employs 6-base oligos hybridised to 100 kilobase DNA fragments. The single stranded DNA is threaded through a nanopore where the hybridised probes are detected by current changes. The software builds the entire sequence of the DNA fragment by deconvoluting the spacing of multiple independent 6-mer hybridisation experiments. NABsys plans to offer whole human genome sequencing for under USD 100. The technologies employing nanopore devices do not modify the native DNA strand and may be collectively referred to as Fourth Generation. The resulting processes are simpler and sequencing more accurate, with the potential to sequence the entire human genome in less than a day. The technical breakthrough in developing nanopore-based sequencing technology will be in controlling the motion of the DNA as it passes through the opening for accurate readings.
The question that still needs to be answered is: which platform should my company invest in or utilise for routine applications now? Because sample preparation and machine run times are similar, one useful principle is to choose platforms that suit current or foreseeable application needs. For de novo sequencing – especially for sequences with repeat regions, where long reads are usually desirable, the 454 platform offers the longest reads. For transcriptomics, chromosome immuno – precipitation and epigenetic studies, shorter reads are sufficient, and can be reliably addressed on Helicos, AB SOLiD and Illumina machines. (These recommendations will, of course, change when the advances in third generation sequencers are fully realised).
The reliability and data output standardisation of these sequencing platforms are currently the focus of a two year study led by the US Food and Drug Administration18. The Sequence Quality Control Initiative (SEQC) distributed identical samples to be sequenced by facilities worldwide employing different sequencing platforms. The data output was then distributed again to various centers using diverse analytical software. Due for publication at the end of 2010, the SEQC report will represent what may be the most extensive study of its kind, and will help develop better standardised protocols, especially for industries where sequence data is used for diagnostics.
The analysis of the first human genome draft naturally led to the question of the genetic relationship between hereditary differences and the onset of disease. In the last decade, various groups have mapped known variable regions among disease-linked and normal populations using microarray based methods in copy number variation (CNV) and genome wide association studies (GWAS)19. It is largely accepted that these studies have failed to adequately identify genetic disease markers, and that the real genetic links may lie not in common variants but in rare alleles. The resolution of individuals’ genomes and transcriptomes down to single nucleotides by sequencing may be necessary to isolate the alleles that explain the molecular basis of disease.
The next step, profiling at the genetic level, could advance medical care by identifying persons likely to benefit most from a particular medication and those most at risk of adverse reactions. Most drugs fail in Phase II of clinical trials, where the drug is tested on a small cohort of patients with the disease. Yet subgroups within the cohort who react positively or negatively to the drug may share genetic markers predictive of such responses. Isolating these markers would allow for the selection of the best responders for testing in the Phase III trials – resulting in better targeted, and therefore safer, more effective drugs. It is tempting to speculate that many compounds that failed in previous clinical trials can be rescued by correcting the patient stratification.
Clinical trials are the most costly part of drug development. Stratifying drug responders earlier in the process (i.e., the pre-clinical stage), will make clinical trials more efficient and less expensive. Most pharmacology studies employ animal models that unavoidably exhibit variable responses during pre-clinical testing. Some models may not be suitable because they differ unpredictably from humans. For example, primate models used for toxicology studies could be of mixed genetic backgrounds because of species inter-breeding, confounding the results of the study with a wide range of drug responses. Sequencing of each test animal will soon become a routine way to identify early markers of traits that influence a drug’s effects, giving an early window on the target population for testing in clinical trials.
Accompanying dramatic decreases in cost, increasing accessibility to sequencing tech – nologies has been a boon to evolutionary research. The full genomic sequences of 3,800 organisms, including 14 unique mammals, have already been deposited in public data archives. The number of human beings sequenced is also increasing: thus far, about 30 human genomes are available to the public in a database maintained by the Personal Genome Project (PGP), whose goals include the collection of 100,000 genomes to allow researchers to catalogue human genetic variation20.
Although few groups have the PGP’s capacity to maintain sequencing data at the rate that it is being produced, several large scale efforts have been founded to sequence smaller human and human-related samples. To name just a few, the Human Microbiome Project aims to sequence all microbial organisms in our bodies21, the Cancer Genome Atlas will map cancer cell lines and primary tumours22, and the Encode/modEncode project will attempt to catalogue all epigenetic markers and DNA binding protein sites in humans and selected model organisms23. Handling the flood of data remains the next biggest challenge in the field. The National Institutes of Health (NIH) and European Bioinformatics Institute (EBI) are gearing up to make their Sequence Reads Archive (SRA) the repository of all raw genome data24. These sequence reads represent the evidence trail for all sequence assemblies, including rare sequences or alleles that might otherwise go undetected. For smaller groups considering warehousing sample data, long term storage may not be an option, as digital storage costs will soon outweigh sequencing costs. Before long, assuming that data analysis tools can keep pace with the rapid changes in sequencing technologies, it will be less expensive to re-sequence a sample than to archive the data for a long period.
Despite all these considerations, though, the most disruptive development in this field will not be a new sequencing platform, storage solution or analytical software package – it will be the deciphering of our genetic code, which will allow us to scale down our efforts and to focus, finally, on the information in our genes that really matters.
Acknowledgement
I am indebted to Chris Schultis and Dr. Jatinderpal Deol for critical reading of the manuscript.
References
1. Sturtevant AH, Third group of linked genes in Drosophila ampelophila, Science 1913, PMID: 17833164
2. Watson JD and Crick FH, the structure of nucleic acids; a structure for deoxyribose nucleic acid, Nature 1953, PMID: 13054692
3. Min Jou W et al,Nucleotide sequence of the gene coding for the bacteriophage MS2 coat protein. Nature 1972, PMID 4555447
4. Sanger F et al, DNA sequencing with chain-terminating inhibitors, PNAS (1977) PMID 271968.
5. Lander ES et al, Initial sequencing and analysis of the human genome, Nature 2001, PMID 11237011
6. Venter C et al, The sequence of the human genome, Science 2001, PMID 11181995
7. Roche 454 (http://www.454.com/)
8. illumina Solexa System (http://www.illumina.com/technology/ sequencing_technology.ilmn)
24. Sequence Reads Archives (SRA) (http://www.ncbi.nlm.nih.gov/sra and http://www.ebi.ac.uk/ena/about/ page.php?page=sra_submissions )
About the Author
Bhupinder Bhullar
Bhupinder Bhullar is a Lab Head in the Novartis Institutes for BioMedical Research (NIBR) in Basel, Switzerland. He obtained his PhD from the University of Calgary, Canada and completed postdoctoral studies at Harvard Medical School and the Whitehead Institute, MIT. Bhupinder’s interest is to develop creative methods for drug discovery to bring novel targets and small molecules into the development pipeline. Bhupinder’s group employs sequencing to study genetic variation and elucidate a drug compound’s mechanism of action.
This website uses cookies to enable, optimise and analyse site operations, as well as to provide personalised content and allow you to connect to social media. By clicking "I agree" you consent to the use of cookies for non-essential functions and the related processing of personal data. You can adjust your cookie and associated data processing preferences at any time via our "Cookie Settings". Please view our Cookie Policy to learn more about the use of cookies on our website.
This website uses cookies to improve your experience while you navigate through the website. Out of these cookies, the cookies that are categorised as ”Necessary” are stored on your browser as they are as essential for the working of basic functionalities of the website. For our other types of cookies “Advertising & Targeting”, “Analytics” and “Performance”, these help us analyse and understand how you use this website. These cookies will be stored in your browser only with your consent. You also have the option to opt-out of these different types of cookies. But opting out of some of these cookies may have an effect on your browsing experience. You can adjust the available sliders to ‘Enabled’ or ‘Disabled’, then click ‘Save and Accept’. View our Cookie Policy page.
Necessary cookies are absolutely essential for the website to function properly. This category only includes cookies that ensures basic functionalities and security features of the website. These cookies do not store any personal information.
Cookie
Description
cookielawinfo-checkbox-advertising-targeting
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertising & Targeting".
cookielawinfo-checkbox-analytics
This cookie is set by GDPR Cookie Consent WordPress Plugin. The cookie is used to remember the user consent for the cookies under the category "Analytics".
cookielawinfo-checkbox-necessary
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
This cookie is set by GDPR Cookie Consent WordPress Plugin. The cookie is used to remember the user consent for the cookies under the category "Performance".
PHPSESSID
This cookie is native to PHP applications. The cookie is used to store and identify a users' unique session ID for the purpose of managing user session on the website. The cookie is a session cookies and is deleted when all the browser windows are closed.
viewed_cookie_policy
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
zmember_logged
This session cookie is served by our membership/subscription system and controls whether you are able to see content which is only available to logged in users.
Performance cookies are includes cookies that deliver enhanced functionalities of the website, such as caching. These cookies do not store any personal information.
Cookie
Description
cf_ob_info
This cookie is set by Cloudflare content delivery network and, in conjunction with the cookie 'cf_use_ob', is used to determine whether it should continue serving “Always Online” until the cookie expires.
cf_use_ob
This cookie is set by Cloudflare content delivery network and is used to determine whether it should continue serving “Always Online” until the cookie expires.
free_subscription_only
This session cookie is served by our membership/subscription system and controls which types of content you are able to access.
ls_smartpush
This cookie is set by Litespeed Server and allows the server to store settings to help improve performance of the site.
one_signal_sdk_db
This cookie is set by OneSignal push notifications and is used for storing user preferences in connection with their notification permission status.
YSC
This cookie is set by Youtube and is used to track the views of embedded videos.
Analytics cookies collect information about your use of the content, and in combination with previously collected information, are used to measure, understand, and report on your usage of this website.
Cookie
Description
bcookie
This cookie is set by LinkedIn. The purpose of the cookie is to enable LinkedIn functionalities on the page.
GPS
This cookie is set by YouTube and registers a unique ID for tracking users based on their geographical location
lang
This cookie is set by LinkedIn and is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
lidc
This cookie is set by LinkedIn and used for routing.
lissc
This cookie is set by LinkedIn share Buttons and ad tags.
vuid
We embed videos from our official Vimeo channel. When you press play, Vimeo will drop third party cookies to enable the video to play and to see how long a viewer has watched the video. This cookie does not track individuals.
wow.anonymousId
This cookie is set by Spotler and tracks an anonymous visitor ID.
wow.schedule
This cookie is set by Spotler and enables it to track the Load Balance Session Queue.
wow.session
This cookie is set by Spotler to track the Internet Information Services (IIS) session state.
wow.utmvalues
This cookie is set by Spotler and stores the UTM values for the session. UTM values are specific text strings that are appended to URLs that allow Communigator to track the URLs and the UTM values when they get clicked on.
_ga
This cookie is set by Google Analytics and is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. It stores information anonymously and assign a randomly generated number to identify unique visitors.
_gat
This cookies is set by Google Universal Analytics to throttle the request rate to limit the collection of data on high traffic sites.
_gid
This cookie is set by Google Analytics and is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visited in an anonymous form.
Advertising and targeting cookies help us provide our visitors with relevant ads and marketing campaigns.
Cookie
Description
advanced_ads_browser_width
This cookie is set by Advanced Ads and measures the browser width.
advanced_ads_page_impressions
This cookie is set by Advanced Ads and measures the number of previous page impressions.
advanced_ads_pro_server_info
This cookie is set by Advanced Ads and sets geo-location, user role and user capabilities. It is used by cache busting in Advanced Ads Pro when the appropriate visitor conditions are used.
advanced_ads_pro_visitor_referrer
This cookie is set by Advanced Ads and sets the referrer URL.
bscookie
This cookie is a browser ID cookie set by LinkedIn share Buttons and ad tags.
IDE
This cookie is set by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
li_sugr
This cookie is set by LinkedIn and is used for tracking.
UserMatchHistory
This cookie is set by Linkedin and is used to track visitors on multiple websites, in order to present relevant advertisement based on the visitor's preferences.
VISITOR_INFO1_LIVE
This cookie is set by YouTube. Used to track the information of the embedded YouTube videos on a website.