Physiologicaly based pharmacokinetic modelling of transporters in drug discovery and development
22
SHARES
Posted: 3 September 2012 |
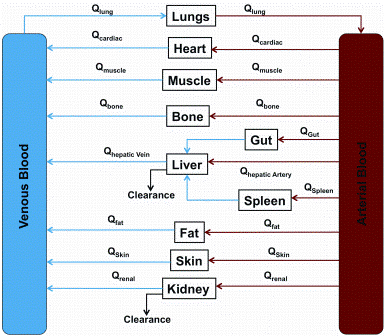
Physiologically based pharmacokinetic (PBPK) models describe the different compartments (tissues) in the body linked via arterial and venous blood flow (Figure 1). The volume of each tissue and blood flows are available from literature data1-5 and PBPK models have been developed for many species including rat, mouse, dog, pig and human2,6,7. PBPK models can be applied to many aspects of the drug develop ment continuum, from drug discovery8 and into development including use in regulatory responses9.
PBPK modelling is becoming a tool of choice in the pharmaceutical industry for the prediction of pharmacokinetic parameters, drugdrug interactions (DDI) and tissue distribution from in vitro data. PBPK modelling was able to become a mainstream tool in the pharma – ceutical industry with advances in in vitro metabolism techniques along with the ability to predict tissue distribution parameters or Kp values for a number of classes of compounds10-13. These models usually assume that the liver and kidney are the only organs where elimination occurs and that blood flow to these organs limits the excretion rate. Recently, with advances in in vitro techniques to study transporter proteins, the input of these data in PBPK models is becoming more commonplace.
Figure 1: Schematic representation of body compartments in physiologically based pharmacokinetic modelling. Abbreviations: Qorgan: Blood flow to organ
Physiologically based pharmacokinetic (PBPK) models describe the different compartments (tissues) in the body linked via arterial and venous blood flow (Figure 1). The volume of each tissue and blood flows are available from literature data1-5 and PBPK models have been developed for many species including rat, mouse, dog, pig and human2,6,7. PBPK models can be applied to many aspects of the drug develop ment continuum, from drug discovery8 and into development including use in regulatory responses9.
This webinar showcases the Growth Direct System; an RMM (Rapid Microbial Method) that improves on traditional membrane filtration, delivering increased accuracy, a faster time to result, enhanced data integrity compliance, and more control over the manufacturing process.
Key learning points:
Understand the benefits of full workflow microbiology quality control testing automation in radiopharmaceutical production
Learn about ITM’s implementation journey and considerations when evaluating the technology
Find out how the advanced optics and microcolony detection capabilities of Growth Direct® technology impact time to result (TTR).
Don’t miss your chance to learn from experts in the industry –Register for FREE
Can’t attend live? No worries – register to receive the recording post-event.
PBPK modelling is becoming a tool of choice in the pharmaceutical industry for the prediction of pharmacokinetic parameters, drugdrug interactions (DDI) and tissue distribution from in vitro data. PBPK modelling was able to become a mainstream tool in the pharma – ceutical industry with advances in in vitro metabolism techniques along with the ability to predict tissue distribution parameters or Kp values for a number of classes of compounds10-13. These models usually assume that the liver and kidney are the only organs where elimination occurs and that blood flow to these organs limits the excretion rate. Recently, with advances in in vitro techniques to study transporter proteins, the input of these data in PBPK models is becoming more commonplace. Drug transporters can be divided into two major classes; efflux and influx. Efflux transporters such as P-glycoprotein and multi-resistance proteins (MRPs) are driven by ATP, pumping drug molecules from tissues, while influx transporters facilitate the entry of polar molecules into the tissues where they are expressed; for example, organic anion transport protein 1B1 (OATP1B1) is responsible for the entry of HMG-CoA reductase inhibitors or ‘statins’ into hepatocytes.
Figure 1: Schematic representation of body compartments in physiologically based pharmacokinetic modelling. Abbreviations: Qorgan: Blood flow to organ
Generally, there are two types of PBPK models, permeability rate limited models which are applicable to describe the pharmacokinetics of compounds that cannot readily passively diffuse across lipid membranes into tissues, while perfusion (i.e. blood flow) limited PBPK models are applicable for compounds that can readily pass into tissues. BDDCS classification14 defines that so-called Class 3 and 4 drugs have low membrane permeability14 and many have been shown to be substrates of transporter proteins. It is therefore recognised that for many compounds in these classes, more traditional perfusion limited PBPK models are not app ropriate for such compounds15.
Absorption models
Absorption of a drug from the gastrointestinal tract into the systemic circulation after oral administration is a multi-step phenomenon, which is influenced by various parameters such as a drug’s physiochemical properties (pKa, lipophilicity, molecular weight, polar surface area, conformation, solubility), formulation parameters (dosage form, crystal form, particle size) as well as the physiology of the human body (gastric emptying time, GI tract surface area, transporters, drug metabolising enzymes)16. Modelling of drug absorption is, therefore, multi-factorial and a complicated nexus of events (Figure 2) and several approaches had been described in the literature (Table 1) to amalgamate these factors.
Table 1: Different approaches for PBPK modelling of drug absorption; the involvement of transporters and their interplay with drug metabolizing enzymes (DMEs)
The metabolism of drugs, as well as their transport across the biological membranes of the intestine and the liver, are two important determinants in oral drug absorption. Small intestine and liver express Phase I and Phase II metabolic enzymes together with uptake and efflux transporters (Figure 2). Traditionally, drug metabolism and transport processes had been considered in the pharmaceutical industry as separate concepts and it is only recently that there has been a paradigm shift to adapt a holistic view to integrate metabolism and transport data in light of emerging evidences that these have been shown to act in tandem to alter the absorption of drugs.
Figure 2: Schematic representation of different steps involved in absorption of drugs on peroral administration along with processes associated at each step and corresponding theoretical / mathematical concept for PBPK modelling of drug absorption. Abbreviations: CYPs : Cytochrome P450, GSTs: Glutathione Sulfotransferases, SULTs: Sulphate transferase, UGTs: UDP-glucuronosyltransferases, MDR1: Mulidrug resistance protein, MRPs: Mulidrug resistance associated protein, BCRP: Breast cancer resistance protein, PEPT: Peptide transporter, OATP: Organic anion-transporting polypeptide, OCT: Organic cation transporter, OCTN: Carinitine/organic cation transporter, ENT: Equilibrative nucleoside transporter, MCT1: Monocarboxylic acid, ASBT: Apical sodium-dependent bile acid transporter
Several attempts have been made to describe the PBPK modelling of transporters and metabolism in the literature (Table 1) and these usually involve biochemical parameters like intrinsic transport clearances, intrinsic metabolic clearances, plasma protein binding, blood to plasma ratio and Michaelis- Menten kinetics. Major challenges in this area are paucity of information about the regional expression of transporters and metabolic enzymes in intestine and liver as well as need to characterise in vitro assays more thoroughly to allow better transformation of data generated in vitro.
PBPK modelling of absorption has the potential to benefit drug discovery and development during pre-clinical and clinical development through its ability to predict absorption from in vitro data, the amount absorbed from various regions of the GI tract, plasma concentrations or drug absorbed as a function of time, the possibility of drug-drug interactions due to biopharmaceutic reasons and mechanistic explanations for observed atypical pharmacokinetic characteristics of drugs (e.g. non-linear PK due to intestinal metabolism and/or transport). Incorporation of metabolism and transport in absorption modelling has greatly enhanced predictions performed by classical models by bringing more reliable estimates in systemic bioavailability, allowing assessment of the effects of efflux and influx transporters in gut or liver, fit complex non-linear metabolism and transport, tracking as well as multiple metabolites and predicting DDIs due to transporters and drug metabolising enzymes. Variability in drug absorption in populations due to genetic polymorphism or ethnic differences in transporters and metabolising enzymes can well be explicable by PBPK.
Liver models
The recent trend within the pharmaceutical industry to develop more polar molecules in an attempt to limit toxicity, reduce P450 mediated metabolic clearance and also make some targets such as ion channels and peptidic receptors accessible means that the likelihood of compounds interacting with transporter proteins has increased over recent years. As a consequence, an ability to predict the hepatic clearance mediated via transporters expressed in the liver has become a significant need, this has begun to be addressed through the development of PBPK models that utilise hydrophilic compounds such as pravastatin as probe substrates6.
Figure 3: OATP: Organic anion-transporting polypeptide, OCT: Organic cation transporter, MDR: Multidrug resistance protein, MRP: Multidrug resistance associated protein, BCRP: Breast cancer resistance protein, NTCP: Sodium taurocholate cotransporting polypeptide, BSEP: Bile Salt Export Pump, MATE: Multidrug and toxin extrusion, CNT: Concentrative nucleoside transporter
As described in Figure 3, both influx (OATPs, OAT2 and OCT1) and efflux (MRP-2, BCRP, MDR1, BSEP) are expressed on the basolateral and cannulicular membranes of human hepatocytes and several PBPK models incorporating the impact of these transporters on the hepatic clearance are available in the literature6,15,17.
PBPK models of transporter mediated hepatic clearance are detailed in Table 2. Watanabe and co-workers6 published one of the earliest PBPK models to describe transporter mediated hepatic clearance. This PBPK model includes both the impact of both influx (OATP1B1) and efflux (MRP2) on the hepatic excretion of pravastatin. This model was further developed by Jones and co-workers17 with several compounds known to be substrates of hepatic transporters using data from sandwich cultured hepatocytes to describe hepatic parameters.
Table 2: PBPK models describing transporter mediated hepatic clearance reported in the literature
Poirier and co-workers15,18 predicted the transporter mediated clearance of the hydrophilic compounds, valsartan, napasagatran and fexofenadine using permeability limited hepatic compartments in PBPK models. In these models, similar to the models described above, the liver was the only tissue considered to be permeability limited. Poirier and co-workers15 also used cell lines expressing organic anion transporting proteins OATP1B1 and OATP1B3 to determine the transporter kinetic parameters of valsartan against both proteins and also relative activity factors (RAF) between the expressed OATP cell lines and human hepatocytes. These data were used to predict the human plasma concentration-time profile and could also be used to simulate the effect of inhibiting one or both of the hepatic uptake proteins.
Kidney models
Figure 3: PBPK models describing renal function reported in the literature
The kidney is one of the most complex physiological organs due to the heterogenity of cell types and the large number of physiological parameters involved in urinary excretion. There are a few reports of PBPK modelling of renal clearance (Table 3) which describe the toxicological effects of environmental pollutants and renal drug pharmacokinetics. Although large number of transporters are expressed on renal tubular cells (Figure 4), there is a scarcity of PBPK models that fully incorporate metabolism and transporter functionalities within them.
Simcyp® software has a module to describe drug pharamcokinetics in renally impaired patients where renal clearance is based on determination of glomerular filtration rate determined from the mathematical relationship between serum creatinine levels and age as well as sex. However, it lacks metabolic and transport mechanisms. As the knowledge of these mechanisms and computational capabilities increases, it is expected that good PBPK renal clearance models will appear in the near future. In a similar perspective, it is noteworthy to highlight two recent publications by the US FDA groups that used renal PBPK modelling. The first publication19 evaluates exposure changes of non-renally eliminated drugs in patients with chronic kidney disease using PBPK modeling as chronic kidney disease or renal impairment (RI) can increase plasma levels for some drugs for whom renal elimination is not a major pathway. Plasma pharmacokinetic profiles of three non-renally eliminated drugs (sildenefil, repaglinide and telithromycin) were simulated in subjects with severe RI and normal renal function and simulated versus observed areas under the concentration versus time curve changes (AUCR, severe RI/normal) were determined. It was observed that simulations correlated better with observed data when the estimated changes in transporter activity due to renal impairment were incorporated into the model. Also, the PBPK models were further used to evaluate the changes in pharmacokinetic profiles of sildenafil metabolite due to RI and of telithromycin due to RI and following coadministration with ketoconazole. The second publication20 from the US FDA was focused on sharing drug review experiences where a PBPK approach to quantitatively predict a complex drug-drug-disease interaction scenario for rivaroxaban during its review process. Quantitative prediction of rivaroxaban exposure was performed in patients with varying degrees of renal impairment when co-administered with a drug that is both a P-gp and CYP3A4 inhibitor. The model provided meaningful mechanistic insights about the possible outcomes and allowed regulators to reach a decision to add cautionary language to the approved product labelling for rivaroxaban.
PBPK and transporter mediated distribution
For compounds that have low intrinsic membrane permeability, transporter proteins can affect their distribution into tissues. Other than tissues that are major drug clearance sites (e.g. liver and kidney), transporters have been shown to effect distribution into tissues, such as brain21, lung22 and heart23. The impact of transporter proteins has been most widely described across the blood-brain barrier24-26 the most well documented example being Pglycoprotein limiting the brain penetration of several drug molecules27-30.
In 2005, Liu and co-workers31 described a simple PBPK model where the brain was described as two compartments, one intracellular and one extracellular, however no active transport was assumed. Four years later, Pglycoprotein was incorporated into PBPK models2 describing a mouse brain, which performed better than a well stirred model. Recently Wagner and co-workers32 described a possible PBPK model of the CNS, incorporating both active uptake and efflux. While this type of model is still to be fully validated, the authors highlight the on-going activities, such as quantification and identification of transporters in the brain that mean a PBPK model describing transporter mediated distribution into the brain will be successfully validated over the next few years. This would be highly impactful, allowing the prediction of such things as concentration-time profiles of drugs, high – lighting species differences in brain penetration and also elucidating the influence of individual transporters on the brain penetration of substrates.
The use of PBPK models to study the impact of transporter proteins on the disposition of drugs in other tissues has not been reported in the literature, probably in part due to the lack of in vitro systems to study transporter activity in tissues other than those listed above. The disposition of a derivative of cyclosporine A5 has been studied in the red blood cells of rats and then inputted into a PBPK model. To adequately describe the distribution of the compound, theoretical transport barriers were included between red blood cells and plasma and also between the unbound compound in tissues and plasma. This enabled the slower than expected tissue distribution into some tissues to be well described.
Regulatory aspects: DDIs, populations and special populations groups
The growing significance of PBPK modelling in regulatory submissions is evident by the fact that from July 2008 to June 2010, the Office of Clinical Pharmacology in the Centre for Drug Evaluation and the Research of the US FDA reviewed seven investigational new drug (IND) and six new drug application (NDA) submissions containing PBPK modelling and simulation conducted by sponsors33. Additionally, recently released US FDA draft guidance on DDIs34 have categorically emphasised PBPK models to be one of the important tools in the prediction of metabolism and/or transporter based DDIs. Their importance is further highlighted by the fact that the US FDA is in the process of preparing a best practices document to facilitate the use of PBPK in regulatory review and labelling35.
The US FDA Clinical pharmacology guidance document on exposure-response relationships36 states “creating a theory or rationale to explain exposure-response relationships through modelling and simulation allows interpolation and extrapolation to better doses and responses in the general population and to subpopulations defined by certain intrinsic and extrinsic factors.” Indeed, the effect of various extrinsic (co-medications, diet, smoking) and intrinsic (age, gender, weight, height, disease, genetic polymorphism, pregnancy, organ dysfunction) factors on the clinical pharmacology of a drug is evaluated during drug development and forms an integral part of drug dossier submissions to regulatory authorities. PBPK models, being mechanistic and dynamic, utilise drug dependent parameters (LogP, pKa, polar surface area, protein binding, metabolism, transport) and system dependent parameters (expression of transporters/enzymes, secretion of gastric acid and bile, urine flow, blood flow rates in organs etc.) to provide useful information in this regard. In particular, specific examples (Table 4) have been cited regarding the use of PBPK to resolve issues pertaining to DDIs due to transporter and / or metabolism, ethnic differences in PK due to genetic polymorphisms of transporters and metabolic enzymes, PK in special patient population groups like renal or hepatic impaired patients and PK in paediatrics or elderly patients.
Table 4: Examples of PBPK models cited in regulatory approval packages obtained from Pharmapendium®(Database version 2012.2) and case studies performed and reported in the literature
Transporter science is growing in knowledge and much still to be explored information is needed to decipher complex clinical drug-drug interactions. Therefore, PBPK modelling of transporter based DDIs remains a challenging area, yet it is growing in interest. It is due to these challenges that the US FDA have quite rightly recommended in their draft guidance on DDIs34 that “sponsors should provide comprehensive justifications on model assumptions, physio – logical and biochemical plausibility, variability and uncertainty measures. The submission containing the use of such advanced models should include a description of the structural model, source and justifications for both systemand drug-dependent parameters, type of error models, model output, data analysis and adequate sensitivity analyses. If predefined models (structural and error) from commercially available software are employed, versions and deviations from the predefined models should be specified. Sponsors are encouraged to communicate with the FDA regarding the use of these models for the prediction of drug-drug interactions.”
Recently, the US FDA undertook a study evaluating the effectiveness of PBPK modelling in dose finding of paediatric patients37. Four recent regulatory submissions made during 2009-2011 for paediatric drug development were reviewed alongside model drugs (midazolam, theophylline, omeprazole) for which PK data was available. It was concluded that there is a critical need to refine paediatric models as PBPK models could not accurately predict body weight normalised clearances for all paediatric groups.
References
1. Jones, H.M., et al., A novel strategy for physiologically based predictions of human pharmacokinetics. Clin.Pharmacokinet., 2006. 45(5): p.511-542
2. Fenneteau, F., et al., Assessing drug distribution in tissues expressing P-glycoprotein through physiologically based pharmacokinetic modeling: model structure and parameters determination. Theor.Biol.Med.Model., 2009. 6 p.2
3. Davies, B. and T. Morris Physiological parameters in laboratory animals and humans. Pharm.Res., 1993. 10(7): p.1093-1095
4. Brown, R.P., et al., Physiological parameter values for physiologically based pharmacokinetic models. Toxicol.Ind.Health, 1997. 13(4): p.407-484
5. Kawai, R., et al., Physiologically based pharmacokinetic study on a cyclosporin derivative, SDZ IMM 125. J.Pharmacokinet.Biopharm., 1994. 22(5): p.327-365
6. Watanabe, T., et al., Physiologically based pharma – cokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J.Pharmacol.Exp.Ther., 2009. 328(2): p.652-662
7. Gustafson, D.L. and D. H. Thamm Pharmacokinetic modeling of doxorubicin pharmacokinetics in dogs deficient in ABCB1 drug transporters. J.Vet.Intern.Med., 2010. 24(3): p.579-586
8. Jones, H.M., I. B. Gardner and K. J. Watson Modelling and PBPK simulation in drug discovery. AAPS J., 2009. 11(1): p.155-166
9. Rowland, M., C. Peck and G. Tucker Physiologicallybased pharmacokinetics in drug development and regulatory science. Annu.Rev.Pharmacol.Toxicol., 2011. 51 p.45-73
10. Poulin, P. and S. Haddad Advancing prediction of tissue distribution and volume of distribution of highly lipophilic compounds from a simplified tissuecomposition- based model as a mechanistic animal alternative method. J.Pharm.Sci., 2012. 101(6): p.2250-2261
11. Rodgers, T., D. Leahy and M. Rowland. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J.Pharm.Sci., 2005. 94(6): p.1259-1276
12. Rodgers, T. and M. Rowland Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J.Pharm.Sci., 2006. 95(6): p.1238-1257
13. Poulin, P. and F. P. Theil Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J.Pharm.Sci., 2002. 91(1): p.129-156
14. Wu, C.Y. and L. Z. Benet Predicting drug disposition via application of BCS: transport/absorption/ elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm.Res., 2005. 22(1): p.11-23
15. Poirier, A., et al., Prediction of pharmacokinetic profile of valsartan in human based on in vitro uptake transport data. J.Pharmacokinet.Pharmacodyn., 2009. 36(6): p.585-611
16. Sharma, P, Chawla, HPS, Panchagnula, R The role of sorption promoters in increasing the bioavailability of drugs in oral preparations. Drugs of the Future, 1999. 24(11): p.1221-1241
17. Jones, H.M., et al., Mechanistic pharmacokinetic modeling for the prediction of transporter-mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab.Dispos., 2012. 40(5): p.1007-1017
18. Poirier, A., et al., Mechanistic modeling of hepatic transport from cells to whole body: application to napsagatran and fexofenadine. Mol.Pharm., 2009. 6(6): p.1716-1733
19. Zhao, P., et al., Evaluation of exposure change of nonrenally eliminated drugs in patients with chronic kidney disease using physiologically based pharmacokinetic modeling and simulation. J.Clin.Pharmacol., 2012. 52(1 Suppl): p.91S-108S
20. Grillo, J.A., et al., Utility of a physiologically-based pharmacokinetic (PBPK) modeling approach to quantitatively predict a complex drug-drug-disease interaction scenario for rivaroxaban during the drug review process: implications for clinical practice. Biopharm.Drug Dispos., 2012. 33(2): p.99-110
21. Begley, D.J. ABC transporters and the blood-brain barrier. Curr.Pharm.Des., 2004. 10(12): p.1295-1312
22. Bosquillon, C. Drug transporters in the lung–do they play a role in the biopharmaceutics of inhaled drugs? J.Pharm.Sci., 2010. 99(5): p.2240-2255
23. Iwata, D., et al., Involvement of carnitine/organic cation transporter OCTN2 (SLC22A5) in distribution of its substrate carnitine to the heart. Drug Metab.Pharmacokinet., 2008. 23(3): p.207-215
24. Loscher, W. and H. Potschka Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx, 2005. 2(1): p.86-98
25. Obradovic, T., et al., Assessment of the first and second generation antihistamines brain penetration and role of P-glycoprotein. Pharm.Res., 2007. 24(2): p.318-327
26. Enokizono, J., et al., Quantitative investigation of the role of breast cancer resistance protein (Bcrp/Abcg2) in limiting brain and testis penetration of xenobiotic compounds. Drug Metab.Dispos., 2008. 36(6): p.995-1002
27. Schinkel, A.H. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv.Drug Deliv.Rev., 1999. 36(2-3): p.179-194.
28. Chen, C., et al., P-glycoprotein limits the brain penetration of nonsedating but not sedating H1- antagonists. Drug Metab.Dispos., 2003. 31(3): p.312-318
29. Muzi, M., et al., Imaging of cyclosporine inhibition of Pglycoprotein activity using 11C-verapamil in the brain: studies of healthy humans. J.Nucl.Med., 2009. 50(8): p.1267-1275
30. Polli, J.W., et al., An unexpected synergist role of Pglycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3- fluorobenzyl)oxy]phenyl}-6-[5-({[2- (methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab.Dispos., 2009. 37(2): p.439-442
31. Liu, X., et al., Use of a physiologically based pharmacokinetic model to study the time to reach brain equilibrium: an experimental analysis of the role of blood-brain barrier permeability, plasma protein binding, and brain tissue binding. J.Pharmacol.Exp.Ther., 2005. 313(3): p.1254-1262
32. Wager, T.T., et al., Strategies to minimize CNS toxicity: in vitro high-throughput assays and computational modeling. Expert Opin.Drug Metab.Toxicol., 2012. 8(5): p.531-542
33. Zhao, P., et al., Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin.Pharmacol.Ther., 2011. 89(2): p.259-267
34. Drug Interaction Studies —Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations.(16 April 2012)
35. Huang, S.M. PBPK as a tool in regulatory review. Biopharm.Drug Dispos., 2012. 33(2): p.51-52 36.
36 Exposure-Response Relationships — Study Design, Data Analysis, and Regulatory Applications. 16 April 2012
37. Leong, R., et al., Regulatory Experience With Physiologically Based Pharmacokinetic Modeling for Pediatric Drug Trials. Clin.Pharmacol.Ther., 2012
38. Agoram, B., W. S. Woltosz and M. B. Bolger Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv.Drug Deliv.Rev., 2001. 50 Suppl 1 p.S41-67
39. Jamei, M., et al., Population-based mechanistic prediction of oral drug absorption. AAPS J., 2009. 11(2): p.225-237
40. Badhan, R., et al., Methodology for development of a physiological model incorporating CYP3A and Pglycoprotein for the prediction of intestinal drug absorption. J.Pharm.Sci., 2009. 98(6): p.2180-2197
41. Cong, D., M. Doherty and K. S. Pang A new physiologically based, segregated-flow model to explain route-dependent intestinal metabolism. Drug Metab.Dispos., 2000. 28(2): p.224-235
42. Gertz, M., J. B. Houston and A. Galetin Physiologically based pharmacokinetic modeling of intestinal firstpass metabolism of CYP3A substrates with high intestinal extraction. Drug Metab.Dispos., 2011. 39(9): p.1633-1642
43. Poirier, A., et al., Design, data analysis, and simulation of in vitro drug transport kinetic experiments using a mechanistic in vitro model. Drug Metab.Dispos., 2008. 36(12): p.2434-2444
44. Meyer, M., et al., Using expression data for quantification of active processes in physiologically based pharmacokinetic modeling. Drug Metab.Dispos., 2012. 40(5): p.892-901
45. Akahane, A. and Kai, M., Konishi, E., Kusama, T., Aoki, Y. Physiologically Based Pharmacokinetics model for estimating urinary excretion of short half-life nuclides in nuclear medicine. Radiation Protection Dosimetry, 1995. 57 p.409-412
46. Corley, R.A., et al., Development of a physiologically based pharmacokinetic model for ethylene glycol and its metabolite, glycolic Acid, in rats and humans. Toxicol.Sci., 2005. 85(1): p.476-490
47. Corley, R.A., G. A. Bormett and B. I. Ghanayem. Physiologically based pharmacokinetics of 2-butoxyethanol and its major metabolite, 2- butoxyacetic acid, in rats and humans. Toxicol.Appl.Pharmacol., 1994. 129(1): p.61-79
48. Russel, F.G., A. C. Wouterse and C. A. van Ginneken. Physiologically based pharmacokinetic model for the renal clearance of phenolsulfonphthalein and the interaction with probenecid and salicyluric acid in the dog. J.Pharmacokinet.Biopharm., 1987. 15(4): p.349-368
49. Boom, S.P., et al., A physiologically based kidney model for the renal clearance of ranitidine and the interaction with cimetidine and probenecid in the dog. Biopharm.Drug Dispos., 1998. 19(3): p.199-208
About the authors
Katherine Fenner is the Associate Director of the Transporter Group, Global DMPK, AstraZeneca. Following com – pletion of a BSc in Chemistry and Pharmacology at the University of Southampton, Katherine Fenner joined the Department of Drug Metabolism at Pfizer in Sandwich. During her 20 year career, Katherine has had experience of many aspects of DMPK science, including pharmacokinetic studies, bioanalysis and in vitro metabolism studies. In the late 1990s, Katherine developed an interest in transporter science, working with Professor Richard Kim at Vanderbilt University at Nashville around OATP transporter proteins. Over the last 10 years, Katherine has worked on many aspects of transporter science, including P-gp mediated drug-drug interactions, prediction of hepatic transporter mediated clearance and hepatic OATP structure-activity. Recent publications include a PBPK model to predict hepatic uptake mediated clearance and the effect of efflux on brain penetration.
Dr Pradeep Sharma is a Senior Scientist in the Transporter Group, Global DMPK at AstraZeneca (UK). He has 10 years of experience working in Johnson and Johnson, AstraZeneca and UCB Pharma in the area of pre-clinical and clinical DMPK in different capacities as study director, study monitor, participating scientist and DMPK project representative. His interests include cellular transport assays, drug metabolism and predictions or simulations of drug-drug interactions. He has a graduate and post-graduate in pharmaceutical sciences from Bangalore University and a PhD in Pharmaceutics (biopharmaceutics) from NIPER. Dr Sharma has 17 publications in peer reviewed international journals, guided two graduate dissertations, 20 published abstracts, one book chapter and is scientific reviewer of drug metabolism journals like Current Drug Metabolism, Drug Metabolism Letters etc.
This website uses cookies to enable, optimise and analyse site operations, as well as to provide personalised content and allow you to connect to social media. By clicking "I agree" you consent to the use of cookies for non-essential functions and the related processing of personal data. You can adjust your cookie and associated data processing preferences at any time via our "Cookie Settings". Please view our Cookie Policy to learn more about the use of cookies on our website.
This website uses cookies to improve your experience while you navigate through the website. Out of these cookies, the cookies that are categorised as ”Necessary” are stored on your browser as they are as essential for the working of basic functionalities of the website. For our other types of cookies “Advertising & Targeting”, “Analytics” and “Performance”, these help us analyse and understand how you use this website. These cookies will be stored in your browser only with your consent. You also have the option to opt-out of these different types of cookies. But opting out of some of these cookies may have an effect on your browsing experience. You can adjust the available sliders to ‘Enabled’ or ‘Disabled’, then click ‘Save and Accept’. View our Cookie Policy page.
Necessary cookies are absolutely essential for the website to function properly. This category only includes cookies that ensures basic functionalities and security features of the website. These cookies do not store any personal information.
Cookie
Description
cookielawinfo-checkbox-advertising-targeting
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertising & Targeting".
cookielawinfo-checkbox-analytics
This cookie is set by GDPR Cookie Consent WordPress Plugin. The cookie is used to remember the user consent for the cookies under the category "Analytics".
cookielawinfo-checkbox-necessary
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
This cookie is set by GDPR Cookie Consent WordPress Plugin. The cookie is used to remember the user consent for the cookies under the category "Performance".
PHPSESSID
This cookie is native to PHP applications. The cookie is used to store and identify a users' unique session ID for the purpose of managing user session on the website. The cookie is a session cookies and is deleted when all the browser windows are closed.
viewed_cookie_policy
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
zmember_logged
This session cookie is served by our membership/subscription system and controls whether you are able to see content which is only available to logged in users.
Performance cookies are includes cookies that deliver enhanced functionalities of the website, such as caching. These cookies do not store any personal information.
Cookie
Description
cf_ob_info
This cookie is set by Cloudflare content delivery network and, in conjunction with the cookie 'cf_use_ob', is used to determine whether it should continue serving “Always Online” until the cookie expires.
cf_use_ob
This cookie is set by Cloudflare content delivery network and is used to determine whether it should continue serving “Always Online” until the cookie expires.
free_subscription_only
This session cookie is served by our membership/subscription system and controls which types of content you are able to access.
ls_smartpush
This cookie is set by Litespeed Server and allows the server to store settings to help improve performance of the site.
one_signal_sdk_db
This cookie is set by OneSignal push notifications and is used for storing user preferences in connection with their notification permission status.
YSC
This cookie is set by Youtube and is used to track the views of embedded videos.
Analytics cookies collect information about your use of the content, and in combination with previously collected information, are used to measure, understand, and report on your usage of this website.
Cookie
Description
bcookie
This cookie is set by LinkedIn. The purpose of the cookie is to enable LinkedIn functionalities on the page.
GPS
This cookie is set by YouTube and registers a unique ID for tracking users based on their geographical location
lang
This cookie is set by LinkedIn and is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
lidc
This cookie is set by LinkedIn and used for routing.
lissc
This cookie is set by LinkedIn share Buttons and ad tags.
vuid
We embed videos from our official Vimeo channel. When you press play, Vimeo will drop third party cookies to enable the video to play and to see how long a viewer has watched the video. This cookie does not track individuals.
wow.anonymousId
This cookie is set by Spotler and tracks an anonymous visitor ID.
wow.schedule
This cookie is set by Spotler and enables it to track the Load Balance Session Queue.
wow.session
This cookie is set by Spotler to track the Internet Information Services (IIS) session state.
wow.utmvalues
This cookie is set by Spotler and stores the UTM values for the session. UTM values are specific text strings that are appended to URLs that allow Communigator to track the URLs and the UTM values when they get clicked on.
_ga
This cookie is set by Google Analytics and is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. It stores information anonymously and assign a randomly generated number to identify unique visitors.
_gat
This cookies is set by Google Universal Analytics to throttle the request rate to limit the collection of data on high traffic sites.
_gid
This cookie is set by Google Analytics and is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visited in an anonymous form.
Advertising and targeting cookies help us provide our visitors with relevant ads and marketing campaigns.
Cookie
Description
advanced_ads_browser_width
This cookie is set by Advanced Ads and measures the browser width.
advanced_ads_page_impressions
This cookie is set by Advanced Ads and measures the number of previous page impressions.
advanced_ads_pro_server_info
This cookie is set by Advanced Ads and sets geo-location, user role and user capabilities. It is used by cache busting in Advanced Ads Pro when the appropriate visitor conditions are used.
advanced_ads_pro_visitor_referrer
This cookie is set by Advanced Ads and sets the referrer URL.
bscookie
This cookie is a browser ID cookie set by LinkedIn share Buttons and ad tags.
IDE
This cookie is set by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
li_sugr
This cookie is set by LinkedIn and is used for tracking.
UserMatchHistory
This cookie is set by Linkedin and is used to track visitors on multiple websites, in order to present relevant advertisement based on the visitor's preferences.
VISITOR_INFO1_LIVE
This cookie is set by YouTube. Used to track the information of the embedded YouTube videos on a website.