G protein coupled receptors – exploiting flexible conformations
6
SHARES
Posted: 3 September 2012 |
G-protein coupled receptors (GPCRs) are a diverse super-family of proteins located within the plasma membrane of eukaryotic cells which have a common architecture consisting of seven-transmembrane (7-TM) segments, connected by extracellular (ECL) and intracellular (ICL) loops. They differ from other 7-TM proteins in their ability to activate guanine-nucleotide binding proteins or β-arrestin and so initiate a signalling cascade. They have a wide range of physiological roles and provide many successful drug targets, playing a role in disorders including allergies, cardiovascular dysfunction, depression, obesity, cancer, pain, diabetes and a variety of central nervous system conditions. This review will give a general overview of GPCRs and how their structures and activities can be used in drug discovery…
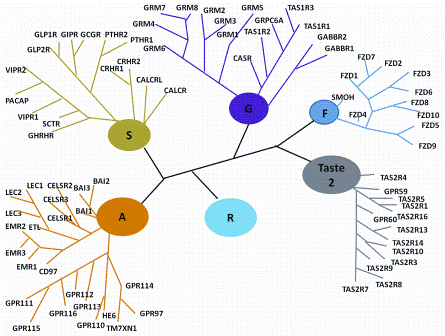
Figure 1: Classification Scheme of GPCRs. R (Rhodopsin-like), S (Secretin-like), G (Glutamate-like), Others (Adhesion, Frizzled, Taste type-2, unclassified)
G-protein coupled receptors (GPCRs) are a diverse super-family of proteins located within the plasma membrane of eukaryotic cells which have a common architecture consisting of seven-transmembrane (7-TM) segments, connected by extracellular (ECL) and intracellular (ICL) loops. They differ from other 7-TM proteins in their ability to activate guanine-nucleotide binding proteins or β-arrestin and so initiate a signalling cascade. They have a wide range of physiological roles and provide many successful drug targets, playing a role in disorders including allergies, cardiovascular dysfunction, depression, obesity, cancer, pain, diabetes and a variety of central nervous system conditions1-3. This review will give a general overview of GPCRs and how their structures and activities can be used in drug discovery.
This webinar showcases the Growth Direct System; an RMM (Rapid Microbial Method) that improves on traditional membrane filtration, delivering increased accuracy, a faster time to result, enhanced data integrity compliance, and more control over the manufacturing process.
Key learning points:
Understand the benefits of full workflow microbiology quality control testing automation in radiopharmaceutical production
Learn about ITM’s implementation journey and considerations when evaluating the technology
Find out how the advanced optics and microcolony detection capabilities of Growth Direct® technology impact time to result (TTR).
Don’t miss your chance to learn from experts in the industry –Register for FREE
Can’t attend live? No worries – register to receive the recording post-event.
GPCRs may be classified into five major families with the Rhodopsin-like being the largest (672 human family members) followed by the Frizzled / Taste (36 members), Adhesion (33), Glutamate (22), and Secretin (15) sub-groups (Figure 1)4. While the Rhodopsin-like family remains the most widely studied, the Adhesion family of GPCRs is particularly fascinating. They have an extended cleavable N-terminal domain thought to be involved in cell-cell contact5 and almost all are still orphan receptors. There are significant associations between the Adhesion group and human disease but as yet, there are no specific small molecules which address their function. Frizzled receptors play a role in governing cell polarity, embryonic development, formation of neural synapses, cell proliferation and many other processes in developing and adult organisms6.
The small Secretin-receptor family is structurally and functionally diverse and includes receptors for polypeptide hormones, and for Drosophila proteins that regulate stress responses and longevity7. Glutamate receptors have a large extracellular N-terminus (often likened to a clam) which binds the orthosteric (endogenous) ligand. Several allosteric ligands to these receptors have been identified and these appear to bind within the seven transmembrane region. Many of the receptors have accessory proteins which either facilitate trafficking to the plasma membrane or influence the specificity of the ligand8.
GPCRs respond to various ligands extracellularly, such as amines, peptides, hydrophobic effectors, hormones, small proteins and volatiles, which allows the receptors to activate the G-proteins intracellularly. Pharmacological agents that act at GPCRs can broadly be categorised into four main classes: agonists, inverse agonists, antagonists and allosteric modulators. Nearly 50 per cent of currently marketed drugs2,9-11 are targeted at less than 20 per cent of these receptors (non-odorant group). This number of drugs is expected to increase as new functions / ligands for GPCRs are discovered and so GPCR targets will remain a very active on-going focus in drug discovery efforts12,13.
Figure 1: Classification Scheme of GPCRs. R (Rhodopsin-like), S (Secretin-like), G (Glutamate-like), Others (Adhesion, Frizzled, Taste type-2, unclassified)
GPCR structure and in silico ligand design
Traditionally, pharmacophore-based app – roaches, where the most relevant structural features of active compounds are collected, have been applied to GPCR ligand design14-16. However, exploitation of the 3D structures of GPCRs is now preferable due to the elucidation of the structure of rhodopsin17, followed by those of the beta118, beta219, adrenergic, adenosine A2A20, dopamine D321, CXCR4 chemokine22, histamine H1 receptors23 and recently sphingosine 1-phosphate receptor24 and the M2 muscarinic acetylcholine receptor25. These GPCRs tend to accommodate their small molecule effectors with similar spatial arrangement, but the particular interactions with amino acid side chains are quite different. These crystal structures have presented the real possibility of using information on the ligandbinding pockets to allow in silico screening for novel small-molecule ligands.
Structure based virtual screening (VS) is typically performed by docking a molecule into the binding site and determining the optimal orientation. Subsequent scoring of these complexes allows assessment of the binding modes and ranking by affinity, prioritisation and biological testing. Carlsson et al26 utilised molecular docking to computationally screen a 1.4 million compound database against the adenosine A2A receptor20. Of those tested, 35 per cent showed substantial activity with affinities between 200 nM and 9 μM with over 50-fold specificity for the adenosine A2A versus the related A1 and A3 adenosine receptor subtypes. Katritch et al27 also screened four million commercially available compounds against the same crystal structure. Out of 56 high ranking compounds tested in adenosine A2A receptor binding assays, 23 showed affinities under 10 μM, 11 of those had sub-μM affinities and two under 60 nM indicating the success of such an approach.
The structures of activated and/or agonistbound GPCRs have also been determined28-31 and can now be utilised in drug design efforts. The biggest structural change observed is a movement outwards from the bilayer of the cytoplasmic parts of the 5th and 6th transmembrane (TM) helices. The structure of activated beta-2 adrenergic receptor in complex with Gs confirmed that the Gα binds to a cavity created by this movement in the vicinity of the 5/6 intracellular loop32.
Homology modelling of GPCRs from X-ray structures is also now more accurate with the use of multi-template models33, the incorporation of knowledge-based constraints and molecular dynamics simulations34-36 and by refining docking poses37. These approaches can indicate potential structural features not revealed by X-ray methods. The feasibility of docking screens against modelled GPCRs was considered in the recent study of Carlsson et al38 on 3.3 million molecules docked into a homology model of the D3 dopamine receptor and subsequently on the released crystal structure. For the homology model, 26 were tested for binding and six had affinities ranging from 0.2 to 3.1 μM. Of the 25 from the crystal structure, five had affinities ranging from 0.3 to 3.0 μM.
GPCR – G protein Assays
Historically, 7-TM receptors were reported to couple to one type of G-protein: e.g. Gi, Gs, Go or Gq, and the method chosen to measure function was linked to the G-protein classification.
Due to the low expression levels of GPCRs in native tissue, the development of recombinant technology has greatly enhanced the way in which we are able to measure GPCR activity. However, this has come at a price. Data from recombinant cell lines can suffer from lack of the appropriate associating proteins, or again, from altered post-translation modification compared to native systems, and thus the receptors can exhibit non-physiological pharmacology39. In addition, recombinant cell lines have high levels of receptor reserve and, therefore, partial agonists or antagonists may appear as full agonists or demonstrate agonism respectively. For instance, N-des-methyl clozapine (NDMC), has been shown to act as a potent partial agonist of the M1 receptor in recombinant systems40-42, but behaves as an antagonist with human tissue43. It is suggested that recombinant systems more efficiently couple to the G-protein, and thus can reach full activation through lower occupancy39,44.
If partial agonists cannot be identified early in drug discovery, this will have an impact on the success of the programme depending on the mode of action required for therapeutic efficacy, since partial agonists will actually lower endogenous agonist response. One successful way of being able to fully characterise full, partial and inverse agonism is by measuring G-protein [35S] GTPγS binding, in preference to reporter gene, calcium flux or downstream kinase assessment assays45. The latter are generally not sensitive enough to report efficacy (Emax) values since they are reliant on accumulation of product, but, [35S] GTPγS binding is a direct measure of GPCR activity.
Recent developments of specific anti-Gs and Gq probes have allowed, in conjunction with scintillation proximity assay (SPA) bead technology46-48, the direct ‘capture’ of Gs and Gq as well as Gi signals49,50 in plate-based formats. These have been used successfully for both recombinant and native tissue systems where knock-out mice and chemical tools are available to validate the system for particular GPCR subtypes50. Recently, label free technologies51 have been developed to measure GPCR efficacy related to specific G-proteins as well as measuring the response in real time, thus capturing all events rather than in a particular time interval assay such as with [35S] GTPγS, where time-dependent events may be missed (reviewed in52).
β-arrestin activity
Label free assays are measures of impedance or optical changes for live cell responses to ligands, which can detect changes in cellular features including adhesion, proliferation, migration and cell death52. GPCRs may not signal by just one process but be functionally selective53. Each transduction system is thought to demonstrate a distinct temporal profile54. For example, agonists may differentially activate G-proteins and/or β-arrestin (biased ligands, Figure 3). Conventionally, the measurement of each event requires a different cell line and cell specific affects cannot be excluded over biased signalling. However, label free technology holds the promise of identifying ligand biased affects in the same cell type. Watts et al55 have demonstrated, for example, CXCL9 as a Gprotein specific agonist of the chemokine receptor CXCR3 compared to previous evidence which suggested a role in desensitisation56. Rational design of such ‘biased’ or ‘functionally selective’ ligands requires a deep structural understanding of the different conformations a GPCR can assume and of how each con – formation influences various downstream G-protein and arrestin signalling pathways – a very challenging objective57.
Figure 3: Currently, GPCRs are considered to utilize two primary types of transducers: G proteins and β- arrestins. Accessory proteins can either facilitate trafficking to the plasma membrane or influence the specificity of the ligand. Homodimerisation, heterodimeristion and transactivation may aid in increasing the diversity of signalling pathways
Allosterism
It has generally been thought that one agonist molecule binds one GPCR molecule; which in turn activates one G-protein subtype. However, gathering evidence suggests that this is not always (or maybe even never has been) the case53. One example is seen with the melanocortin 4 receptor (MC4R) which we have demonstrated forms homodimers when recombinantly expressed in the HEK-293 cell line (unpublished data). When a mutant receptor that lacked the ability to bind the agonist was co-expressed with a receptor that could not signal, MC4R signalling was restored. Such transactivation58 has been further demonstrated in vivo54, whereby, in a transgenic mouse model expressing binding-deficient luteinising hormone receptor (LHR) and signalling deficient LHR, normal signalling occurred59.
Attention now is focusing not only on homo-dimerisation and its functional consequence but also on hetero-dimerisation where such distinct heterodimers should be classed as a molecular target rather than the individual proteins60,61 (Figure 3). Likewise, GPCRs come under a number of allosteric pressures from accessory proteins; for example we have demonstrated that the G-protein negatively modulates agonist binding in the MC4R (unpublished data) and in the absence of the G-protein (high GTPγS concentrations), agonist occupancy increases. Likewise, Secretin GPCRs are well documented to require accessory proteins such as RAMPs for function62,63. This kind of observation, alongside the premise of receptor reserve oligomerisation, cell specific G-protein coupling and accessory protein requirement is driving pharmacological investigation away from simple recombinant systems towards more ‘native-like’ systems for studying GPCR function in the hope of higher success rates in the identification of novel therapeutics.
GPCR pharmacology
Changes in ‘typical’ GPCR pharmacology are also reflected in the types of ligands that are being sought. Identification of allosteric modulation of GPCRs is not novel, but the pharmaceutical search for an alternative mechanism to activate GPCR function has expanded into this space. Positive allosteric modulators have typically enhanced endogenous agonist activity. However, it is as yet unclear whether the ‘alternative’ binding site nevertheless still resides in the penumbra of the agonist site as has been suggested by mutagenesis. The recent interest in dimerisation / complementation and accessory proteins has employed allosterism not only for modulating the orthosteric site, but also protein interactions that modulate GPCR function.
Dualsteric ligands represent a novel mode of targeting GPCRs by addressing simul – taneously both the orthosteric and an allosteric binding site e.g. the design of dualsteric muscarinic agonists64. To date, discovery efforts for several GPCR subtypes have failed to deliver really selective drug candidates65 but potentially, with a dualsteric approach, orthosteric receptor activation may be linked with allosteric subtype-selectivity and intracellular signalling pathway selectivity in a single molecular entity. Such an approach may also simultaneously target more than one GPCR, e.g. bivalent beta2- adrenergic and adenosine A1 receptor ligands66.
Pharmacological chaperones
Mutated versions of membrane proteins can result in local miss-folding, preventing efficient trafficking to the plasma membrane and exposure to circulating ligands. This can result in disease. Effective trafficking can be rescued using pharmacological chaperones (small hydro – phobic molecules that can penetrate the cell membrane) which bind to the nascent GPCR, and presumably restore the native fold. Conn and Janovick67 have pioneered the use of cell-permeant small molecule Gonadotropinreleasing hormone (GnRH) receptor antagonists to rescue poorly expressed GnRH mutant receptors. Recently, a set of MC4R mutants, linked with an obese phenotype, has been successfully rescued to the plasma membrane by the MC4R inverse agonist ML0025376468. To examine the potential effect that these mutations may have on the local receptor structure and the binding poses of the identified pharmacological chaperones with MC4R, we developed a model of the ‘inactive form’ of MC4R and the three mutants V50M, S58C and I137T (unpublished work). From our computational analysis, it is evident that such point mutations can result in changed interactions which may alter helical-helical packing and effect receptor stability and/or activation steps without directly affecting the ligand binding pocket. Additionally, the local change induced with each of the three mutants is different and further supports a hypothesis that each is retained by a different molecular mechanism. Exposure to putative pharmacological chaperones was shown to increase the plasma membrane expression of the wild type and the three mutant MC4Rs, V40M, S58C and I137T (unpublished work).
Conclusions
This short review has attempted to capture the essence of the family of GPCRs as we currently understand them. Increasing knowledge of their structural and functional characteristics has opened up new avenues for intervention strategies for GPCR-related human conditions. In particular, their apparent atypical conformational flexibility has been intriguing, giving rise to the prospect of new therapeutic approaches. At the same time, however, it will continue to be important to develop assay systems that address this dynamic behaviour, not only with respect to different conformational states but also probably different forms of quaternary associations.
References
1. Deshpande DA, Penn RB: Targeting g protein-coupled receptor signaling in asthma. Cell Signal (2006) 18(12):2105-2120
2. Hill SJ: G-protein-coupled receptors: Past, present and future. Br J Pharmacol (2006) 147 Suppl 1(S27-37
3. Catapano LA, Manji HK: G protein-coupled receptors in major psychiatric disorders. Biochim Biophys Acta (2007) 1768(4):976-993
4. Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB: The g-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol (2003) 63(6):1256-1272
5. Aplin AE, Howe A, Alahari SK, Juliano RL: Signal transduction and signal modulation by cell adhesion receptors: The role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacol Rev (1998) 50(2):197-263
6. Huang HC, Klein PS: The frizzled family: Receptors for multiple signal transduction pathways. Genome Biol (2004) 5(7):234
12. Vassilatis DK, Hohmann JG, Zeng H, Li F, Ranchalis JE, Mortrud MT, Brown A, Rodriguez SS, Weller JR, Wright AC, Bergmann JE et al: The g protein-coupled receptor repertoires of human and mouse. Proc Natl Acad Sci U S A (2003) 100(8):4903-4908
13. Kroeze WK, Sheffler DJ, Roth BL: G-proteincoupled receptors at a glance. J Cell Sci (2003) 116 (Pt 24):4867-4869
14. Wei J, Qu W, Ye Y, Gao Q: 3d pharmacophore based virtual screening of a 2a adenosine receptor antagonists. Protein Pept Lett (2010) 17(3):332-339
15. Joseph T, Suneel Kumar B, Santhosh B, Kriti S, Pramod A, Ravikumar M, Kishore M: Quantitative structure activity relationship and pharmacophore studies of adenosine receptor a2b inhibitors. Chem Biol Drug Des (2008) 72(5):395-408
16. Malo M, Brive L, Luthman K, Svensson P: Selective pharmacophore models of dopamine d(1) and d(2) full agonists based on extended pharmacophore features. ChemMedChem (2010) 5(2):232-246
17. Palczewski K, Kumasaka T, Hori T, Behnke C, Motoshima H, Fox B, Le Trong I, Teller D, Okada T, Stenkamp R, Yamamoto M et al: Crystal structure of rhodopsin: A g protein-coupled receptor. Science (2000) 289(5480):739-745
18. Warne T, Serrano-Vega M, Baker J, Moukhametzianov R, Edwards P, Henderson R, Leslie A, Tate C, Schertler G: Structure of a beta1-adrenergic g-protein-coupled receptor. Nature (2008) 454(7203):486-491
19. Rasmussen S, Choi H, Rosenbaum D, Kobilka T, Thian F, Edwards P, Burghammer M, Ratnala V, Sanishvili R, Fischetti R, Schertler G et al: Crystal structure of the human beta2 adrenergic g-protein-coupled receptor. Nature (2007) 450(7168):383-387
20. Jaakola V, Griffith M, Hanson M, Cherezov V, Chien E, Lane J, Ijzerman A, Stevens R: The 2.6 angstrom crystal structure of a human a2a adenosine receptor bound to an antagonist. Science (2008) 322(5905):1211-1217
21. Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC: Structure of the human dopamine d3 receptor in complex with a d2/d3 selective antagonist. Science (2010) 330(6007):1091-1095
22. Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ et al: Structures of the cxcr4 chemokine gpcr with smallmolecule and cyclic peptide antagonists. Science (2010) 330(6007):1066-1071
23. Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T et al: Structure of the human histamine h1 receptor complex with doxepin. Nature (2011) 475(7354):65-70
24. Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, Sanna MG et al: Crystal structure of a lipid g protein-coupled receptor. Science (2012) 335(6070):851-855
25. Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, Kobayashi T: Structure of the human m2 muscarinic acetylcholine receptor bound to an antagonist. Nature (2012) 482(7386):547-551
26. Carlsson J, Yoo L, Gao ZG, Irwin JJ, Shoichet BK, Jacobson KA: Structure-based discovery of a2a adenosine receptor ligands. J Med Chem (2010) 53(9):3748-3755
27. Katritch V, Jaakola V, Lane J, Lin J, Ijzerman A, Yeager M, Kufareva I, Stevens R, Abagyan R: Structure-based discovery of novel chemotypes for adenosine a(2a) receptor antagonists. J Med Chem (2010) 53(4):1799- 1809
28. Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A et al: Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature (2011) 469(7329):175-180
29. Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, Devree BT, Sunahara RK, Chae PS et al: Structure and function of an irreversible agonist-β(2) adrenoceptor complex. Nature (2011) 469(7329):236-240
30. Warne T, Moukhametzianov R, Baker JG, Nehmé R, Edwards PC, Leslie AG, Schertler GF, Tate CG: The structural basis for agonist and partial agonist action on a β(1)-adrenergic receptor. Nature (2011) 469(7329):241-244
31. Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC: Structure of an agonist-bound human a2a adenosine receptor. Science (2011) 332(6027):322-327
32. Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM et al: Crystal structure of the β2 adrenergic receptor-gs protein complex. Nature (2011) 477(7366):549-555
33. Kneissl B, Leonhardt B, Hildebrandt A, Tautermann CS: Revisiting automated g-protein coupled receptor modeling: The benefit of additional template structures for a neurokinin-1 receptor model. J Med Chem (2009) 52(10):3166-3173
34. Jójárt B, Kiss R, Viskolcz B, Keseru GM: Activation mechanism of the human histamine h4 receptor–an explicit membrane molecular dynamics simulation study. J Chem Inf Model (2008) 48(6):1199-1210
35. Rodríguez D, Piñeiro Á, Gutiérrez-de-Terán H: Molecular dynamics simulations reveal insights into key structural elements of adenosine receptors. Biochemistry (2011) 50(19):4194-4208
36. Romo TD, Grossfield A, Pitman MC: Concerted interconversion between ionic lock substates of the beta(2) adrenergic receptor revealed by microsecond timescale molecular dynamics. Biophys J (2010) 98(1):76-84
37. Yarnitzky T, Levit A, Niv MY: Homology modeling of gprotein- coupled receptors with x-ray structures on the rise. Curr Opin Drug Discov Devel (2010) 13(3):317-325
38. Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK: Ligand discovery from a dopamine d3 receptor homology model and crystal structure. Nat Chem Biol (2011) 7(11):769-778
39. Kenakin T: Differences between natural and recombinant g protein-coupled receptor systems with varying receptor/g protein stoichiometry. Trends Pharmacol Sci (1997) 18(12):456-464
40. Sur C, Mallorga PJ, Wittmann M, Jacobson MA, Pascarella D, Williams JB, Brandish PE, Pettibone DJ, Scolnick EM, Conn PJ: N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates n-methyl-daspartate receptor activity. Proc Natl Acad Sci U S A (2003) 100(23):13674-13679
41. Lameh J, Burstein ES, Taylor E, Weiner DM, Vanover KE, Bonhaus DW: Pharmacology of n-desmethylclozapine. Pharmacol Ther (2007) 115(2):223-231
42. Weiner DM, Meltzer HY, Veinbergs I, Donohue EM, Spalding TA, Smith TT, Mohell N, Harvey SC, Lameh J, Nash N, Vanover KE et al: The role of m1 muscarinic receptor agonism of n-desmethylclozapine in the unique clinical effects of clozapine. Psychopharmacology (Berl) (2004) 177(1-2):207-216
43. Thomas DR, Dada A, Jones GA, Deisz RA, Gigout S, Langmead CJ, Werry TD, Hendry N, Hagan JJ, Davies CH, Watson JM: N-desmethylclozapine (ndmc) is an antagonist at the human native muscarinic m(1) receptor. Neuropharmacology (2010) 58(8):1206-1214
44. Nelson CP, Challiss RA: ‘Phenotypic’ Pharmacology: The influence of cellular environment on g protein-coupled receptor antagonist and inverse agonist pharmacology. Biochem Pharmacol (2007) 73(6):737-751
45. Strange PG: Use of the gtpγs ([35s]gtpγs and eu-gtpγs) binding assay for analysis of ligand potency and efficacy at g protein-coupled receptors. Br J Pharmacol (2010) 161(6):1238-1249
46. Cussac D, Newman-Tancredi A, Duqueyroix D, Pasteau V, Millan MJ: Differential activation of gq/11 and gi(3) proteins at 5-hydroxytryptamine(2c) receptors revealed by antibody capture assays: Influence of receptor reserve and relationship to agonist-directed trafficking. Mol Pharmacol (2002) 62(3):578-589
47. DeLapp NW: The antibody-capture [(35)s]gtpgammas scintillation proximity assay: A powerful emerging technique for analysis of gpcr pharmacology. Trends Pharmacol Sci (2004) 25(8):400-401
48. DeLapp NW, McKinzie JH, Sawyer BD, Vandergriff A, Falcone J, McClure D, Felder CC: Determination of [35s]guanosine-5′-o-(3-thio)triphosphate binding mediated by cholinergic muscarinic receptors in membranes from chinese hamster ovary cells and rat striatum using an anti-g protein scintillation proximity assay. J Pharmacol Exp Ther (1999) 289(2):946-955
49. Mannoury la Cour C, Vidal S, Pasteau V, Cussac D, Millan MJ: Dopamine d1 receptor coupling to gs/olf and gq in rat striatum and cortex: A scintillation proximity assay (spa)/antibody-capture characterization of benzazepine agonists. Neuropharmacology (2007) 52(3):1003-1014
50. Salah-Uddin H, Thomas DR, Davies CH, Hagan JJ, Wood MD, Watson JM, Challiss RA: Pharmacological assessment of m1 muscarinic acetylcholine receptorgq/ 11 protein coupling in membranes prepared from postmortem human brain tissue. J Pharmacol Exp Ther (2008) 325(3):869-874
51. Giaever I, Keese CR: Monitoring fibroblast behavior in tissue culture with an applied electric field. Proc Natl Acad Sci U S A (1984) 81(12):3761-3764
52. Scott CW, Peters MF: Label-free whole-cell assays: Expanding the scope of gpcr screening. Drug Discov Today (2010) 15(17-18):704-716
53. Kenakin T: Functional selectivity in gpcr modulator screening. Comb Chem High Throughput Screen (2008) 11(5):337-343
54. Guo W, Shi L, Filizola M, Weinstein H, Javitch JA: Crosstalk in g protein-coupled receptors: Changes at the transmembrane homodimer interface determine activation. Proc Natl Acad Sci U S A (2005) 102(48):17495-17500
55. Watts AO, Scholten DJ, Heitman LH, Vischer HF, Leurs R: Label-free impedance responses of endogenous and synthetic chemokine receptor cxcr3 agonists correlate with g(i)-protein pathway activation. Biochem Biophys Res Commun (2012) 419(2):412-418
56. Colvin RA, Campanella GS, Sun J, Luster AD: Intracellular domains of cxcr3 that mediate cxcl9, cxcl10, and cxcl11 function. J Biol Chem (2004) 279(29):30219-30227
57. Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, Shaw DE: Activation mechanism of the β2- adrenergic receptor. Proc Natl Acad Sci U S A (2011) 108(46):18684-18689
58. Carrillo JJ, Pediani J, Milligan G: Dimers of class a g protein-coupled receptors function via agonistmediated trans-activation of associated g proteins. J Biol Chem (2003) 278(43):42578-42587
59. Rivero-Müller A, Chou YY, Ji I, Lajic S, Hanyaloglu AC, Jonas K, Rahman N, Ji TH, Huhtaniemi I: Rescue of defective g protein-coupled receptor function in vivo by intermolecular cooperation. Proc Natl Acad Sci U S A (2010) 107(5):2319-2324
60. Milligan G, Smith NJ: Allosteric modulation of heterodimeric g-protein-coupled receptors. Trends Pharmacol Sci (2007) 28(12):615-620
61. Smith NJ, Milligan G: Allostery at g protein-coupled receptor homo- and heteromers: Uncharted pharmacological landscapes. Pharmacol Rev (2010) 62(4):701-725
62. Hay DL, Poyner DR, Sexton PM: Gpcr modulation by ramps. Pharmacol Ther (2006) 109(1-2):173-197
63. Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W, Muff R, Fischer JA, Foord SM: International union of pharmacology. Xxxii. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev (2002) 54(2):233-246
64. Mohr K, Tränkle C, Kostenis E, Barocelli E, De Amici M, Holzgrabe U: Rational design of dualsteric gpcr ligands: Quests and promise. Br J Pharmacol (2010) 159(5):997- 1008
65. Conn PJ, Christopoulos A, Lindsley CW: Allosteric modulators of gpcrs: A novel approach for the treatment of cns disorders. Nat Rev Drug Discov (2009) 8(1):41-54
66. Karellas P, McNaughton M, Baker SP, Scammells PJ: Synthesis of bivalent beta2-adrenergic and adenosine a1 receptor ligands. J Med Chem (2008) 51(19):6128-6137
67. Conn PM, Janovick JA: Trafficking and quality control of the gonadotropin releasing hormone receptor in health and disease. Mol Cell Endocrinol (2009) 299(2):137-145
68. Fan ZC, Tao YX: Functional characterization and pharmacological rescue of melanocortin-4 receptor mutations identified from obese patients. J Cell Mol Med (2009) 13(9B):3268-3282
About the authors
Dr Kathryn Chapman is a senior assay development scientist and a member of Imperial College Academic Drug Discovery Centre. She is involved in a number of drug discovery programmes focusing on early hit discovery and pharmacological profiling.
Professor John Findlay studies membrane proteins which are hugely important for investigations ranging from fundamental studies on structure and cellular biology, to humanitarian and commercial applications in areas such as drug discovery and the emergent field of biosensors.
Dr Gemma Kinsella is a Health Research Board (HRB) postdoctoral research fellow in the Membrane Protein Lab, of the Department of Biology, National University of Ireland Maynooth. Her research focuses on protein structure prediction and the early stages of drug development for diseases encompassing a number of GPCR targets.
This website uses cookies to enable, optimise and analyse site operations, as well as to provide personalised content and allow you to connect to social media. By clicking "I agree" you consent to the use of cookies for non-essential functions and the related processing of personal data. You can adjust your cookie and associated data processing preferences at any time via our "Cookie Settings". Please view our Cookie Policy to learn more about the use of cookies on our website.
This website uses cookies to improve your experience while you navigate through the website. Out of these cookies, the cookies that are categorised as ”Necessary” are stored on your browser as they are as essential for the working of basic functionalities of the website. For our other types of cookies “Advertising & Targeting”, “Analytics” and “Performance”, these help us analyse and understand how you use this website. These cookies will be stored in your browser only with your consent. You also have the option to opt-out of these different types of cookies. But opting out of some of these cookies may have an effect on your browsing experience. You can adjust the available sliders to ‘Enabled’ or ‘Disabled’, then click ‘Save and Accept’. View our Cookie Policy page.
Necessary cookies are absolutely essential for the website to function properly. This category only includes cookies that ensures basic functionalities and security features of the website. These cookies do not store any personal information.
Cookie
Description
cookielawinfo-checkbox-advertising-targeting
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertising & Targeting".
cookielawinfo-checkbox-analytics
This cookie is set by GDPR Cookie Consent WordPress Plugin. The cookie is used to remember the user consent for the cookies under the category "Analytics".
cookielawinfo-checkbox-necessary
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
This cookie is set by GDPR Cookie Consent WordPress Plugin. The cookie is used to remember the user consent for the cookies under the category "Performance".
PHPSESSID
This cookie is native to PHP applications. The cookie is used to store and identify a users' unique session ID for the purpose of managing user session on the website. The cookie is a session cookies and is deleted when all the browser windows are closed.
viewed_cookie_policy
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
zmember_logged
This session cookie is served by our membership/subscription system and controls whether you are able to see content which is only available to logged in users.
Performance cookies are includes cookies that deliver enhanced functionalities of the website, such as caching. These cookies do not store any personal information.
Cookie
Description
cf_ob_info
This cookie is set by Cloudflare content delivery network and, in conjunction with the cookie 'cf_use_ob', is used to determine whether it should continue serving “Always Online” until the cookie expires.
cf_use_ob
This cookie is set by Cloudflare content delivery network and is used to determine whether it should continue serving “Always Online” until the cookie expires.
free_subscription_only
This session cookie is served by our membership/subscription system and controls which types of content you are able to access.
ls_smartpush
This cookie is set by Litespeed Server and allows the server to store settings to help improve performance of the site.
one_signal_sdk_db
This cookie is set by OneSignal push notifications and is used for storing user preferences in connection with their notification permission status.
YSC
This cookie is set by Youtube and is used to track the views of embedded videos.
Analytics cookies collect information about your use of the content, and in combination with previously collected information, are used to measure, understand, and report on your usage of this website.
Cookie
Description
bcookie
This cookie is set by LinkedIn. The purpose of the cookie is to enable LinkedIn functionalities on the page.
GPS
This cookie is set by YouTube and registers a unique ID for tracking users based on their geographical location
lang
This cookie is set by LinkedIn and is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
lidc
This cookie is set by LinkedIn and used for routing.
lissc
This cookie is set by LinkedIn share Buttons and ad tags.
vuid
We embed videos from our official Vimeo channel. When you press play, Vimeo will drop third party cookies to enable the video to play and to see how long a viewer has watched the video. This cookie does not track individuals.
wow.anonymousId
This cookie is set by Spotler and tracks an anonymous visitor ID.
wow.schedule
This cookie is set by Spotler and enables it to track the Load Balance Session Queue.
wow.session
This cookie is set by Spotler to track the Internet Information Services (IIS) session state.
wow.utmvalues
This cookie is set by Spotler and stores the UTM values for the session. UTM values are specific text strings that are appended to URLs that allow Communigator to track the URLs and the UTM values when they get clicked on.
_ga
This cookie is set by Google Analytics and is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. It stores information anonymously and assign a randomly generated number to identify unique visitors.
_gat
This cookies is set by Google Universal Analytics to throttle the request rate to limit the collection of data on high traffic sites.
_gid
This cookie is set by Google Analytics and is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visited in an anonymous form.
Advertising and targeting cookies help us provide our visitors with relevant ads and marketing campaigns.
Cookie
Description
advanced_ads_browser_width
This cookie is set by Advanced Ads and measures the browser width.
advanced_ads_page_impressions
This cookie is set by Advanced Ads and measures the number of previous page impressions.
advanced_ads_pro_server_info
This cookie is set by Advanced Ads and sets geo-location, user role and user capabilities. It is used by cache busting in Advanced Ads Pro when the appropriate visitor conditions are used.
advanced_ads_pro_visitor_referrer
This cookie is set by Advanced Ads and sets the referrer URL.
bscookie
This cookie is a browser ID cookie set by LinkedIn share Buttons and ad tags.
IDE
This cookie is set by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
li_sugr
This cookie is set by LinkedIn and is used for tracking.
UserMatchHistory
This cookie is set by Linkedin and is used to track visitors on multiple websites, in order to present relevant advertisement based on the visitor's preferences.
VISITOR_INFO1_LIVE
This cookie is set by YouTube. Used to track the information of the embedded YouTube videos on a website.