Status and challenges in structure-based drug discovery for G protein-coupled receptors
6
SHARES
Posted: 13 December 2011 |
The central location of G protein-coupled receptors (GPCRs) at the interface between the interior and exterior of cells, as well as their key role in signalling events, make GPCRs a prominent class of pharmaceutical targets. To date, approximately 40 per cent of known drugs are thought to act on GPCRs either directly or indirectly. GPCRs are for the most part inaccessible to structural determination due to difficulties to express, purify and crystallise them; however, progress of structure determination has led to seven new structures in the last decade. This number is still insufficient to conduct structure-based drug discovery on all available targets. Computational modelling is therefore a very useful surrogate and in this paper I discuss the reliability of atomistic three-dimensional models that are obtained through molecular modelling in light of the GPCRdock 2008 and 2010 competitions organised by the Scripps Institute. G protein coupled receptors (GPCRs) are key proteins involved in signalling and as such are prominent drug targets. Ligands that bind to GPCRs include small aminergic neuro – transmitters or hormones such as noradrenaline and adrenaline, dopamine, histamine, small peptides, nucleic acids, lipids or even opsins that contain light-reactive retinal chromophores. Altogether, in the human genome project, about 390 non-olfactory GPCRs have been identified; of which about 100 are orphan proteins without an identified ligand or cellular function…
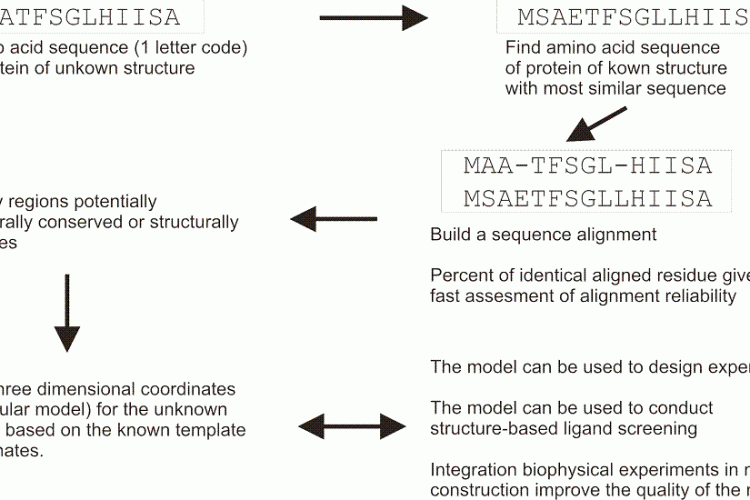
FIGURE 1 Schematic overview of a typical homology modelling procedure that is used to build threedimensional coordinates for a protein of unknown structure
The central location of G protein-coupled receptors (GPCRs) at the interface between the interior and exterior of cells, as well as their key role in signalling events, make GPCRs a prominent class of pharmaceutical targets. To date, approximately 40 per cent of known drugs are thought to act on GPCRs either directly or indirectly. GPCRs are for the most part inaccessible to structural determination due to difficulties to express, purify and crystallise them; however, progress of structure determination has led to seven new structures in the last decade. This number is still insufficient to conduct structure-based drug discovery on all available targets. Computational modelling is therefore a very useful surrogate and in this paper I discuss the reliability of atomistic three-dimensional models that are obtained through molecular modelling in light of the GPCRdock 2008 and 2010 competitions organised by the Scripps Institute.
This webinar showcases the Growth Direct System; an RMM (Rapid Microbial Method) that improves on traditional membrane filtration, delivering increased accuracy, a faster time to result, enhanced data integrity compliance, and more control over the manufacturing process.
Key learning points:
Understand the benefits of full workflow microbiology quality control testing automation in radiopharmaceutical production
Learn about ITM’s implementation journey and considerations when evaluating the technology
Find out how the advanced optics and microcolony detection capabilities of Growth Direct® technology impact time to result (TTR).
Don’t miss your chance to learn from experts in the industry –Register for FREE
Can’t attend live? No worries – register to receive the recording post-event.
G protein coupled receptors (GPCRs) are key proteins involved in signalling and as such are prominent drug targets1. Ligands that bind to GPCRs include small aminergic neuro – transmitters or hormones such as noradrenaline and adrenaline, dopamine, histamine, small peptides, nucleic acids, lipids or even opsins that contain light-reactive retinal chromophores. Altogether, in the human genome project, about 390 non-olfactory GPCRs have been identified; of which about 100 are orphan proteins without an identified ligand or cellular function.
In cells, GPCRs are embedded in lipidic membranes, leading these proteins to adopt a fold composed of seven transmembrane helices, hence the names 7TM or serpentine receptors. GPCRs are critical in mediating signalling events, where ligand binding to the receptor leads to a conformational switch in its structure, activation of an intracellular G-protein and triggering of second messenger pathways. This key location at the interface between inside and outside the cells makes GPCRs validated or emerging drug targets. To date, approximately 40 per cent of marketed drugs are thought to target GPCRs directly or indirectly2 and individual GPCRs are often seen as useful targets for several therapeutic areas. For example, α2-adrenoceptors are thought to be useful targets for the treatment of elevated blood pressure and intraocular pressure, in alleviating withdrawal symptoms from opioid and in alcohol abuse, as well as anesthetic adjuvants. It is therefore of no surprise that GPCRs are of key interest to the pharmaceutical industry.
Ligand-based methodologies in drug discovery relate to the methods that use as starting material the structure of a known ligand and look for more potent or chemically accessible derivatives. Drugs acting at least partially via GPCRs, such as morphine, ephedrine or mescaline, have been documented for a long time. Endogenous chemicals able to activate GPCRs, such as the chromophore 11-cis-retinal or the small molecules such as adrenaline were identified at the start of the 19th century but their mode of action via GPCRs was clarified only much later. In addition to chance observation, drug molecules have been derived from the chemical structure of known molecules, for example the β-blocker propanolol and the antihistaminic cimetidine, which were synthesised in the 1960’s by Sir James Black, recipient of the Nobel prize in 1988 for that work.
A classical approach in modern ligandbased design is to use a pharmacophoric description, or chemical substructures, as starting points to screen in silico commercial or private compound libraries. The last two decades have seen the development of Selective Optimisation of Side Activities (SOSA) that aims at finding new targets for old drugs, and this strategy has shown itself to be very useful with GPCRs that hold many side activities3. More recently, fragment based methodologies have been used for GPCRs too4. A potential drawback of ligand-based methods is that they usually suggest molecules that resemble the initial chemical scaffold.
Structure-based GPCR drug discovery
A more ambitious strategy in drug design is to use information from the atomistic threedimensional atomic structure of the target protein. This strategy requires knowledge of the atomic coordinates for the receptor. In terms of drug discovery, receptor structure-based drug design has its own advantage since it is more likely to result in an entirely novel chemical scaffold than ligand-based methods.
A common description of the molecular mechanism of ligand binding is given by the lock-and-key classical analogy, although a more modern conception is the description of a handin- glove that better represents the flexibility of the interaction partners. A large number of GPCR ligands bind within a pocket located within the plane of the helical transmembrane bundle5. This binding pocket, also called binding cavity, is faced by amino acids that mirror the physicochemical properties of the ligand molecules, allowing an optimal network of weak molecular interactions to take place. The event of ligand binding is complicated to study, due for example to errors in model structures, to the dynamics of the receptor, to water molecules that associate to both ligand and receptor, bridging polar groups of ligand and generating salvation / desolvation costs that are difficult to measure accurately.
The key step to conduct structure-based drug discovery is therefore to obtain the three dimensional coordinates for protein itself. While the first GPCR, rhodopsin, was sequenced in 1975, followed by the adrenergic receptor in the 1980s, it is only in the last decade that reliable structural information has become available for this family. GPCRs are embedded in the cell membrane and have proven themselves to be very resilient to X-ray crystallography, being very difficult to express, solubilise, and crystallise. Rhodopsin, expressed in the eye at high concentration naturally, was the first GPCR structure to be solved at atomic resolution in 20006, although the 3D electronic density of the helices had been determined by cryo-electron microscopy in the 1990s. The human β2-adrenoceptor7 and the turkey β1-adrenoceptor8, the dopamine 3 receptor9, the chemokine CXCR410, and the histamine 1 receptor11 were solved following major break – throughs in the field of X-ray crystallography: ligand-affinity chromatography for purification, robotic systems for microcrystallisation, or e.g. stabilisation by fusion with the T4 lyzosyme protein in place of the third intracellular12,13.
In the absence of crystal structures, molecular models are a widely used surrogate to conduct structure-based drug discovery. Molecular modelling is inexpensive and for GPCRs often the only accessible strategy to gain an atomistic understanding in the ligand binding properties. Molecular modelling aims both to predict the three dimensional structure of proteins and, with molecular docking simulations, to predict the energetically optimal virtual fit of a ligand inside a binding pocket. Docking simulations are also used for prioritisation of compound libraries, i.e. to rank compounds, through virtual screening experiments14.
Methods to build three-dimensional atomic models of GPCRs
Amino acid sequences have been the first and most accessible type of data available for GPCRs, and amino acid sequences are imprinted with information about the three-dimensional structure. Early methods have accurately predicted the seven helical structure based on hydropathy plots, hydrophobic moment plots have resolved for most part the rotational orientation of the helices, while receptor orientation has been successfully predicted using the positive-inside rule. The simple topological models derived from these methods have been very useful to help predict which amino acids are forming the binding pocket, which in turn were used to design mutated receptors to validate the predictions.
The first glimpse of the seven-trans – membrane helical GPCR structure came from cryo-EM maps, which were used to fit experimental data and to build low resolution 3D models in combination with sequencebased prediction methods. Bacteriorhodopsin, a bacterial proton pump predicted to have seven helices and covalently bound with a retinal chromophore, rhodopsin, was first thought to share similarities with known GPCRs and was used early on for structural modelling15. Nonetheless, only the threedimensional structure of bovine rhodopsin really opened new avenues in the field, serving as a basis for a wealth of studies based on homology modelling.
Homology modelling is based on the hypothesis that related proteins share a common fold. In homology modelling (Figure 1), a known protein structure is used as a structural template to build the threedimensional coordinates of an unknown one. A pairwise sequence alignment is used to define the equivalent amino acids. The alignment may allow the user to identify structurally conserved regions, well aligned, and structurally variables regions, often poorly aligned. The protein fold is shared within structurally conserved regions, but the structurally variables regions needs to be modelled independently of the structural template. A commonly used benchmark to represent the evolutionary closeness and therefore the accuracy of the model is a percentage of sequence identity: two random sequences have a sequence identity at most of 15 per cent, and GPCRs share usually about 18-20 per cent identical residues in their transmembrane regions, while receptors for example among the amine family will share 40 per cent. In GPCR modelling to date, the main computational challenge is to find and allow productive deviations from the atomic coordinates inside the structurally conserved regions, as well as in the independent modelling of the structurally variable regions.
FIGURE 1 Schematic overview of a typical homology modelling procedure that is used to build threedimensional coordinates for a protein of unknown structure
State-of-the-art in GPCR complexes modelling: GPCRdock 2008 and GPCRdock 2010
The Scripps Institute has recently organised benchmarking competitions, where the participating groups were asked before the release of the three-dimensional coordinates to build structural models of the adenosine 2A receptor (GPCRdock 2008)16, and of the dopamine 3 and the chemokine CXCR4 receptors (GPCRdock 2010)17, as well as their interaction with ligands. A maximum of one month was given to conduct the modelling study and five to ten models at maximum could be submitted for each group, after that the X-ray structure was released. This type of experiment presents a very accurate evaluation of the pitfalls and of the successes achievable in the field.
A major finding of these studies is that homology modelling followed by docking can be relatively accurate when structure and template share above 35 per cent of identical amino acid, but human knowledge plays an important role: only the best research groups were able to recreate atomic resolution complexes in the most accessible experiment (Figure 2). Generally, the experiment confirmed that the transmembrane helices are nearly always well modelled but for some specific local deviations such as kinks. On the other hand, it is not possible to date to construct accurate models of the loop region of the receptors in a robust and reliable way. Alternatives to homology modelling, for example MemSTRUK, TASSER, GPCRfold, were shown to be in constant improvement, however they did not fare as well as homology modelling. Docking the ligand appeared to be a major challenge. In this type of double-blind study, as in real cases, if the modelling of the receptor structure is wrong, for example if the binding site has been partly obstructed by loops, it impacts the simulation by preventing the successful docking of ligands.
FIGURE 2 Near-atomic resolution modeling of Eticlopride binding to the Dopamine 3 receptor. Selected amino acids lining the binding pocket and their surfaces are shown for the crystal structure (a and c) and for the computational model proposed by myself (b and d) to the GPCRdock 2010 competition. Two key interactions, a hydrogen-bond and salt bridge, are indicated by a green dashed line
The GPCRdock 2010 proposed three targets: the dopamine 3 receptor, the chemokine CXCR4 receptor in complex with a peptide and a small molecule. The dopamine 3 receptor, which is a relatively close relative to the β-adrenoceptor, was well modelled in complex with eticlopride by about one third of the groups. Using experimental information and a sound chemical knowledge appeared key to success. The second target proved itself very challenging and although some groups found the general location of the peptide on the receptor, close to the extracellular surface, no group managed to get an atomistic picture of the molecular interactions. In GPCRdock 2008, modelling of the adenosine 2A receptor with the ligand was also very challenging, with only one group succeeding in building a reasonable model. The successful group had long-lasting experience in working with the adenosine 2A receptor and the SAR series of ligand, showing a clear connection between human knowledge and the ability to build successful GPCR-ligand complexes.
Summary
In summary, computer simulations have proven to be invaluable tools to provide structural information on GPCRs and their interaction with ligands. This type of method still has a brilliant future due to the high pharmaceutical importance of these receptors as drug targets and to the relative inaccessibility of GPCRS to structure determination – although the last decade has seen many improvements in that field. The helical region of GPCRs is usually conserved and well modelled. The reliability of the molecular models can be predicted using simple metrics such as sequence identity. For distant receptors, accurate atomistic modelling is not accessible although these models can be useful to design experiments. For receptor sharing more 35 per cent sequence identity to template, recent blind modelling competitions have shown that it is possible to obtain reliable atomistic models, but only when a wealth of pharmacological and biophysical data is integrated in the molecular models.
About the Author
Henri Xhaard is principal investigator at the Centre for Drug Research, Faculty of Pharmacy, University of Helsinki, Finland where he leads the Computational Drug Discovery group. He received his PhD in 2006 from the University of Åbo Akademi, Finland, after a MSc in molecular biophysics conducted at the University Paris VI. His main expertise is computational modelling of G protein-coupled receptors and he has been ranked among the world top groups in the recent GPCRdock 2010 competition organised by the Scripps institute. The current research of his group includes, in addition to computational drug discovery, pharmacokinetic predictions using QSAR modelling, and development of computational tools to accelerate drug discovery.
References
1. Lundstrom KH, Chiu ML, editors. G protein-coupled receptors in drug discovery. CRC Press; 2006
2. Overington JP, Al-Lazikani B., Hopkins AL. How many drug targets are there. Nature Reviews Drug Discovery 2006; 5:993-996
3. Allen JA, Roth BL. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2011; 51:117-144
4. Congreve M, Langmead C, Marshall FH. The use of GPCR structures in drug design. Adv Pharmacol. 2011;62:1-36
5. Mouillac B, Chini B, Balestre MN, Elands J, Trumpp- Kallmeyer S, Hoflack J, Hibert M, Jard S, Barberis C. The binding site of neuropeptide vasopressin V1a receptor. Evidence for a major localization within transmembrane regions J Biol Chem. 1995;270:25771-7.
6. Palczewski K, Kumasaka T, Hori T, Behnke C, Motoshima H, Fox B, Trong I, Teller D, Okada T, Stenkamp R, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000. 289:739-745
7. Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science.200, 318, 1258-65
9. Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091-5
10. Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with smallmolecule and cyclic peptide antagonists. Science.010;330:1066-71
11. Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011, 475:65-70
12. Landau EM, Pebay-Peroula E, Neutze R. Structural and mechanistic insight from high resolution structures of archeal rhodopsin. FEBS letter 2003; 555: 51-56
13. Cherezov V. Peddi A, Muthusubramanian L, Zheng YF, Caffrey M. A robotic system for crystallizing membrane and soluble proteins in lipidic mesophases. Acta Crystallogr 2004: D60: 1795-1807
14. Bissantz C, Bernard P, Hibert M, Rognan D. Proteinbased virtual screening of chemical databases. II. Are homology models of G-Protein Coupled Receptors suitable targets? Proteins. 2003;50:5-25
15. Trumpp-Kallmeyer S, Hoflack J, Bruinvels A, Hibert M. Modelling of G-protein-coupled receptors: application to dopamine, adrenaline, serotonin, acetylcholine, and mammalian opsin receptors. J Med Chem. 1992;35:3448-62
16. Michino M, Abola E; GPCR Dock 2008 participants, Brooks CL 3rd, Dixon JS, Moult J, Stevens RC. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat Rev Drug Discov. 2009:455-63
17. Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R; GPCR Dock 2010 participants. Status of GPCR modelling and docking as reflected by communitywide GPCR Dock 2010 assessment. Structure. 2011;19:1108-26
This website uses cookies to enable, optimise and analyse site operations, as well as to provide personalised content and allow you to connect to social media. By clicking "I agree" you consent to the use of cookies for non-essential functions and the related processing of personal data. You can adjust your cookie and associated data processing preferences at any time via our "Cookie Settings". Please view our Cookie Policy to learn more about the use of cookies on our website.
This website uses cookies to improve your experience while you navigate through the website. Out of these cookies, the cookies that are categorised as ”Necessary” are stored on your browser as they are as essential for the working of basic functionalities of the website. For our other types of cookies “Advertising & Targeting”, “Analytics” and “Performance”, these help us analyse and understand how you use this website. These cookies will be stored in your browser only with your consent. You also have the option to opt-out of these different types of cookies. But opting out of some of these cookies may have an effect on your browsing experience. You can adjust the available sliders to ‘Enabled’ or ‘Disabled’, then click ‘Save and Accept’. View our Cookie Policy page.
Necessary cookies are absolutely essential for the website to function properly. This category only includes cookies that ensures basic functionalities and security features of the website. These cookies do not store any personal information.
Cookie
Description
cookielawinfo-checkbox-advertising-targeting
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertising & Targeting".
cookielawinfo-checkbox-analytics
This cookie is set by GDPR Cookie Consent WordPress Plugin. The cookie is used to remember the user consent for the cookies under the category "Analytics".
cookielawinfo-checkbox-necessary
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
This cookie is set by GDPR Cookie Consent WordPress Plugin. The cookie is used to remember the user consent for the cookies under the category "Performance".
PHPSESSID
This cookie is native to PHP applications. The cookie is used to store and identify a users' unique session ID for the purpose of managing user session on the website. The cookie is a session cookies and is deleted when all the browser windows are closed.
viewed_cookie_policy
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
zmember_logged
This session cookie is served by our membership/subscription system and controls whether you are able to see content which is only available to logged in users.
Performance cookies are includes cookies that deliver enhanced functionalities of the website, such as caching. These cookies do not store any personal information.
Cookie
Description
cf_ob_info
This cookie is set by Cloudflare content delivery network and, in conjunction with the cookie 'cf_use_ob', is used to determine whether it should continue serving “Always Online” until the cookie expires.
cf_use_ob
This cookie is set by Cloudflare content delivery network and is used to determine whether it should continue serving “Always Online” until the cookie expires.
free_subscription_only
This session cookie is served by our membership/subscription system and controls which types of content you are able to access.
ls_smartpush
This cookie is set by Litespeed Server and allows the server to store settings to help improve performance of the site.
one_signal_sdk_db
This cookie is set by OneSignal push notifications and is used for storing user preferences in connection with their notification permission status.
YSC
This cookie is set by Youtube and is used to track the views of embedded videos.
Analytics cookies collect information about your use of the content, and in combination with previously collected information, are used to measure, understand, and report on your usage of this website.
Cookie
Description
bcookie
This cookie is set by LinkedIn. The purpose of the cookie is to enable LinkedIn functionalities on the page.
GPS
This cookie is set by YouTube and registers a unique ID for tracking users based on their geographical location
lang
This cookie is set by LinkedIn and is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
lidc
This cookie is set by LinkedIn and used for routing.
lissc
This cookie is set by LinkedIn share Buttons and ad tags.
vuid
We embed videos from our official Vimeo channel. When you press play, Vimeo will drop third party cookies to enable the video to play and to see how long a viewer has watched the video. This cookie does not track individuals.
wow.anonymousId
This cookie is set by Spotler and tracks an anonymous visitor ID.
wow.schedule
This cookie is set by Spotler and enables it to track the Load Balance Session Queue.
wow.session
This cookie is set by Spotler to track the Internet Information Services (IIS) session state.
wow.utmvalues
This cookie is set by Spotler and stores the UTM values for the session. UTM values are specific text strings that are appended to URLs that allow Communigator to track the URLs and the UTM values when they get clicked on.
_ga
This cookie is set by Google Analytics and is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. It stores information anonymously and assign a randomly generated number to identify unique visitors.
_gat
This cookies is set by Google Universal Analytics to throttle the request rate to limit the collection of data on high traffic sites.
_gid
This cookie is set by Google Analytics and is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visited in an anonymous form.
Advertising and targeting cookies help us provide our visitors with relevant ads and marketing campaigns.
Cookie
Description
advanced_ads_browser_width
This cookie is set by Advanced Ads and measures the browser width.
advanced_ads_page_impressions
This cookie is set by Advanced Ads and measures the number of previous page impressions.
advanced_ads_pro_server_info
This cookie is set by Advanced Ads and sets geo-location, user role and user capabilities. It is used by cache busting in Advanced Ads Pro when the appropriate visitor conditions are used.
advanced_ads_pro_visitor_referrer
This cookie is set by Advanced Ads and sets the referrer URL.
bscookie
This cookie is a browser ID cookie set by LinkedIn share Buttons and ad tags.
IDE
This cookie is set by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
li_sugr
This cookie is set by LinkedIn and is used for tracking.
UserMatchHistory
This cookie is set by Linkedin and is used to track visitors on multiple websites, in order to present relevant advertisement based on the visitor's preferences.
VISITOR_INFO1_LIVE
This cookie is set by YouTube. Used to track the information of the embedded YouTube videos on a website.