

Committee for orphan medicines – overview of 2013 and looking ahead
Posted: 5 February 2014 | | No comments yet
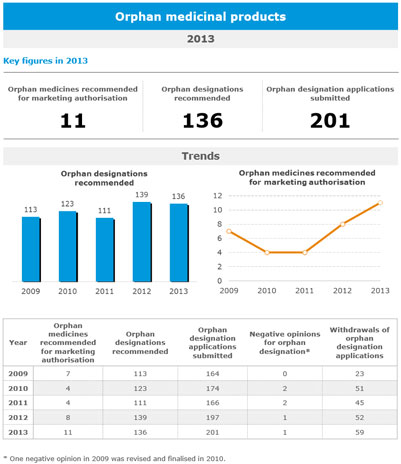
A total of 11 out of 81 medicines recommended for marketing authorisation by the European Medicines Agency’s Committee for Medicinal Products for Human Use in 2013 were intended for the treatment of rare diseases…

A total of 11 out of 81 medicines recommended for marketing authorisation by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) in 2013 were intended for the treatment of rare diseases. This continues the year-on-year increase in the number of marketing authorisation recommendations for medicines for rare diseases observed over the last few years (with 8 in 2012 and 4 in 2011).
 One of the explanations offered by the Committee for Orphan Medicinal Products (COMP), the EMA committee responsible for recommending orphan designation of medicines for rare diseases, is that developers of orphan medicines are now making much better use of the tools offered by the Agency and the European Orphan Regulation to support them in the development of their medicines.
One of the explanations offered by the Committee for Orphan Medicinal Products (COMP), the EMA committee responsible for recommending orphan designation of medicines for rare diseases, is that developers of orphan medicines are now making much better use of the tools offered by the Agency and the European Orphan Regulation to support them in the development of their medicines.
“12 years after the European Orphan Regulation came into force, it is undoubtedly serving its purpose, with more and more orphan medicines reaching patients with rare diseases each year,” explains Bruno Sepodes, Chair of the COMP. “The joint efforts of the EMA committees, including the COMP, the Paediatric Committee and the CHMP, in providing scientific advice to companies during the development of their medicines, have been a driving force behind this success.”
The volume of applications for orphan designation submitted to the Agency has increased by more than 22% since 2009. Moreover, the number of companies seeking protocol assistance (scientific advice for sponsors developing orphan medicines) during the development of their medicines increased from 82 requests in 2012 to 108 requests in 2013. Seeking and following scientific advice has been proven to significantly increase the probability of a positive outcome for marketing authorisation applications.
In 2013, the CHMP recommended granting conditional marketing authorisations for four orphan medicines, a higher number than in previous years (with one in 2012 and none in 2011). This trend is mirrored by an increase in the number of companies requesting advice on the conditional marketing authorisation procedure compared with previous years. This mechanism allows the granting of marketing authorisations on the basis of less complete data for certain categories of medicines, such as orphan medicines, in order to meet unmet medical needs of patients and in the interests of public health.
Micro, small or medium-sized enterprises (SME) are very active in the development of orphan medicines. Approximately 40% of the applications for an orphan designation are submitted by SMEs. The Agency encourages companies developing orphan medicines to register as an SME. These companies benefit from further incentives, including regulatory, administrative and procedural assistance from the Agency’s SME office as well as fee reductions.
“In 2014, we will continue to review and adapt our internal procedures. This will help us to better meet the needs of patients, with whom the Committee has been working in close contact since its creation, and to keep up-to-date with the scientific knowledge on rare diseases. Another priority will be to continue to actively contribute to scientific publications and scientific debate in our area,” adds Bruno Sepodes.
Facilitating access to medicines
The question of reimbursement of orphan medicines once they have received a marketing authorisation is crucial to allow medicines to reach patients.
Orphan medicines represent one of the key areas of collaboration in the EMA-EUnetHTA three-year work plan 2013-2015. The Agency is working with EUnetHTAExternal link icon, an organisation that focuses on scientific cooperation in health technology assessment (HTA) in Europe, towards a better understanding of the specific characteristics of orphan medicines, their marketing authorisation and national competent authorities’ initiatives on availability of designated orphan medicines.
In addition, two medicines with an orphan designation have already been through the Agency’s programme of parallel scientific advice with HTA bodies. These procedures allowed the developers to get advice from the CHMP on the demonstration of a positive benefit-risk balance of their medicine, from the COMP on the establishment of a significant benefit brought by the medicine and from HTA bodies on the demonstration of relative effectiveness.
In 2014, the COMP intends to further develop the concept of parallel scientific advice.
The committee is also planning to raise awareness of the need for designated orphan medicines to demonstrate a significant benefit in order to obtain 10-year market exclusivity, a major incentive provided by the Orphan Regulation. Developers can request protocol assistance for advice on how to demonstrate a significant benefit. In 2013, the COMP received 28 requests for protocol assistance related to the demonstration of significant benefit.
International collaboration
The EMA and the United States Food and Drug Administration (FDA) started working together in 2008 to encourage applications for orphan designation to be submitted in parallel to the two agencies. This programme was extended to the Japanese regulatory authorities (MHLW and PMDA) in 2012.
This programme has been very successful so far. One in two applications for orphan designation was submitted in parallel to other regulators in 2013 and a similar trend is expected in 2014.
The parallel submission process helps companies to rationalise the development of orphan medicines by facilitating access to parallel scientific advice from the three regulatory authorities.
As part of this international collaboration, the three agencies intend to focus their efforts in 2014 on the post-designation programmes offered in the three territories.
In March 2014, the EMA, the FDA and the Japanese regulatory authorities will organise a joint workshop at the EMA office to provide comprehensive information on the orphan programmes in the three territories for orphan medicines developers and offer the opportunity for the participants to present a draft application for orphan designation and discuss issues with the three agencies.
About the Orphan Regulation and the COMP
The Orphan Regulation was introduced in the European Union (EU) to incentivise the development of medicines for rare diseases. Orphan designation can be granted at any stage of the development of medicines intended for diagnosis, prevention or treatment of life-threatening or very serious conditions that affect not more than 5 in 10,000 people in the EU or for which development costs would not be covered by the marketing return without incentives. If methods of diagnosis, prevention or treatment of the disease already exist, orphan designation is only possible if there is a significant benefit to patients offered by the medicine. Sponsors of medicines designated as orphan medicines by the European Commission benefit from a range of incentives, including market exclusivity for 10 years, fee-reductions for some of the Agency’s services, protocol assistance and access to the centralised authorisation procedure.
The COMP is the committee at the EMA that is responsible for reviewing applications from people or companies seeking orphan designation.
Next week, the EMA will publish an overview of 2013 and priorities in 2014 for its Committee for Advanced Therapies (CAT).
