Improving aseptic connections throughout the manufacturing process
Posted: 22 July 2024 | Vanessa Vasadi Figueroa (VVF Science) | No comments yet
As companies are adapting to EU GMP guidelines for Annex 1 – Manufacture of Sterile Medicinal products, there have been numerous discussions involving the terms sterile and aseptic, as if they were synonymous. Vanessa Vasadi Figueroa, Chief Microbiologist at VVF Science® and Executive Director of Microbiology & Sterility Assurance at Quality Executive Partners, Inc, explains that this is not correct, and gives a microbiologist’s perspective on why the distinction between these two terms is important to the larger conversation about sterility assurance.

The great benefit of the recent EU Annex 1 update, is that it enforced the concepts for contamination control and the responsibility for its cross-functional implementation.1 However, this necessitates correct use and application of key terminology such as aseptic and sterile.
Aseptic indicates a high probability that the process is free from contamination. The term aseptic is neither absolute nor precise, and it is not accurate to use the term to describe entire rooms, areas or facilities. Aseptic is best applied to smaller, discrete techniques, behaviours and/or processes, eg, aseptic manufacturing and aseptic process simulation.
Second, the more critical term of the two, is sterile – a stronger word that indicates something is absolutely free of microorganisms, which can be measured using a validated process. This term is best applied to drug substance or product, or items rendered sterile using a validated process such as sterile filtration. It is not accurate to use ‘sterile’ to describe surfaces, methodologies, or environments such as biological safety cabinets and isolators. The term ‘sterile’ is not to be used casually.
The very linkages between each manufacturing control lacks proper understanding and awareness of aseptic techniques. The process of connecting utilities (eg, water for injection) to manufacturing equipment – ie, connecting the sterilising filter to a product tank or responding to leaks in a single-use system – has become more important than ever. As we evolve into more precise processing for biologics or advanced therapy medicinal products (ATMPs), these manual aseptic connections will become more crucial in meeting critical process parameters for microbiological quality.
Release of sterile medicinal products – looking at the focal points
To understand the contamination requirements for which a process needs to be controlled, is to first consider product quality attributes. Not all processes require finished product sterility, so we must be judicious in understanding the process and applying a risk-based approach to the requirements for bioburden control.2 Also, not all bioburden is created equal (endotoxins are byproducts of bacteria that can remain in a solution even after terminal sterilisation),3 bioburden is not uniformly distributed in a solution, and large quantities of bioburden cannot effectively be removed from the process without consequence.4 For example, sterilising-grade filters have upper limits before clogging or other phenomena occur.
Upstream of the process, the controls are less stringent, ie, bioburden-controlled conditions, whereas downstream, they evolve to require more strict conditions of low bioburden, perhaps ~10 CFU/100mL at the pre-filtration stage. The conditions under which bioburden is controlled should be defined in a contamination control strategy (CCS), but these can depend on some key factors5 including the management and quality of materials used to connect equipment, as well as the type of closure levels6 needed when managing exposure of the process to its surrounding environment.
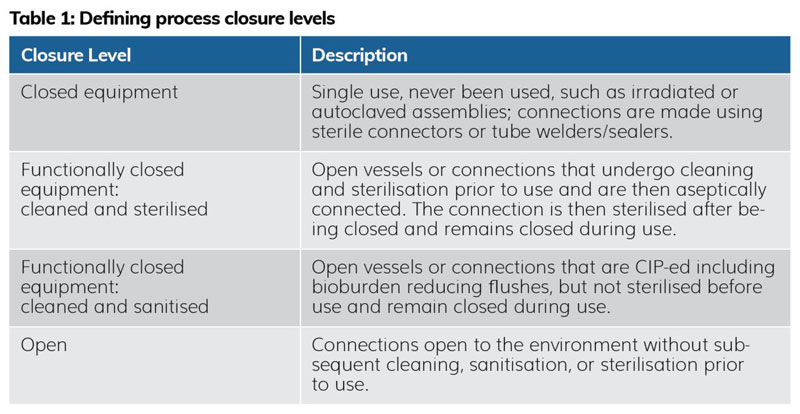
When managing closure levels, it is important for microbiologists and sterility assurance SMEs to understand the many states of ‘closure’ that exist on the manufacturing floor, to support development of the CCS and advise on reasonable areas of risk mitigation. As illustrated in Table 1,6 a process can have fully closed equipment, functionally closed equipment (two types), and open equipment. Open connections are exposed to the environment without subsequent cleaning, sanitisation, or sterilisation steps prior to use, thus pose contamination risk to the process. Bacteria, yeasts and moulds are opportunists and can contaminate the process where it is least expected.
credit: Vanessa Vasadi Figueroa
Therefore, interrogating the equipment, the process and any technology used for performing connections is a first step. Identifying the possibility of bringing closure to the open connection or establishing that it must remain open, then incorporating best practices for improving the way the connection is made, should be the next step. Investigations have resulted in root causes where contamination occurred during an open connection, or when manipulations took place over an open vessel despite downstream controls. The overall number of connections and total length of tubing required to make a connection should also be minimised. If connections must remain open, microbiology and manufacturing departments must work together to align on proper aseptic techniques and behaviours for appropriately managing the conditions of making the connection.
An aseptic connection [will] minimise any potential for contamination, not achieve sterility
There are several considerations when managing open connections. During setup, an operator should know their remote environment where the connection is to be made, which can sometimes be challenging, eg, in tight spaces. This requires some mapping of the task to gather materials, parts, utensils and any gowning/PPE that is needed to perform the connection in advance. It is important to ensure that the area in which materials are set down or handled is cleaned or is amenable to sanitisation. Tubing or parts should be visually inspected to ensure quality before being installed; eg, verifying package integrity, sterilisation cycle indicators and ensuring bonnets or muffs are secured on the end of tubing. A stainless-steel table or cart, and even a secondary operator to facilitate the aseptic connection, can be very useful too. To establish the cleanest boundary, all materials should be sterile and all surfaces should be properly sanitised. Operators must also plan for abandoning any dropped processing materials, thus should bring backup gaskets, clamps or tubing assemblies.
sterile [is] a stronger word that indicates something is absolutely free of microorganisms, which can be measured using a validated process
Once setup is complete, the operator should prepare for execution. Everyone defines “minimal, short or small duration of time” differently, so performing the actual open connection should be timed in countable seconds. It is called an “aseptic connection” because the requirement is to minimise any potential for contamination, not achieve sterility. These same processing materials should not touch the floor, a wall or the gowned person handling them during the live connection. It is better to use new parts than to risk contaminating a formulation or other downstream processing vessel. Aseptic technique also demands that we never directly touch the surface of a gasket, clamp system, tubing assembly ends, or swipe hands over top of an open vessel. An insider tip from microbiologists is to use the outer packaging of sterilised materials/parts to handle or manipulate the part into its final position. It is not possible to achieve sterile conditions for an open connection, but this does not mean an ‘anything goes’ mentality is acceptable. Careful consideration for contamination controls and collaboration between microbiology and manufacturing can lead to fewer risks to product quality.
About the author
Vanessa Vasadi Figueroa is Chief Microbiologist at VVF Science®, and Executive Director of Microbiology & Sterility Assurance at Quality Executive Partners, Inc. With almost 20 years combined experience in the life sciences industry, Figueroa’s core expertise is rooted in sterility assurance, contamination control, microbial investigations, environmental and utilities monitoring programmes and quality control laboratory management.
She has a bachelor’s degree in molecular biology from State University of New York at New Paltz, and a master’s degree in molecular biology and microbiology from San Jose State University. She is an active and publishing member of several professional organisations, such as the Parenteral Drug Association, American Society of Microbiology and Association for Women in Science.
References
- European Commission. Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use: Annex 1 Manufacture of Sterile Medicinal Products.The Rules Governing Medicinal Products in the European Union, 22 Aug. 2022, Brussels, C (2022) 5938 final.
- United States Pharmacopeial Convention. Bioburden Control of Nonsterile Drug Substances and Products.United States Pharmacopeia, General Chapter <1115>, Dec. 2014.
- Removal of Endotoxin from Protein in Pharmaceutical Processes. [Internet] American Pharmaceutical Review. 2016. [cited 2024June]. Available from: americanpharmaceuticalreview.com/Featured-Articles/190810-Removal-of-Endotoxin-from-Protein-in-Pharmaceutical-Processes/.
- Jornitz MW, et al. Considerations in Sterile Filtration-Part II: The Sterilizing Filter and Its Organism Challenge, A Critique of Regulatory Standards. PDA Journal of Pharmaceutical Science and Technology, BioPhorum Operations Group (BPOG).
- Bain D, et al. Microbial Monitoring for Biological Drug Substance Manufacturing: An Industry Perspective. PDA Journal of Pharmaceutical Science and Technology, vol. 69, no. 3, 2015, pp. 451-460. BioPhorum Operations Group (BPOG).
- International Society for Pharmaceutical Engineering. Baseline Guide, Vol. 6: Biopharmaceutical Manufacturing Facilities. 3rd ed, ISPE, 2022.