NITROSAMINES: Where now?
Posted: 28 February 2023 | Dave Elder (David P Elder Consultancy) | No comments yet
While determining active intakes and control strategies for short chain alkyl N-nitrosamines was relatively easy to establish, nitrosamine drug substance-related impurities continue to present challenges. Here, Dave Elder reflects on the three main instances of nitrosamine contamination in drug products.
IN THE INITIAL PHASE of the N-nitrosamines crisis, the issue was the formation of small alkyl chain N-nitrosamines, eg, N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA) (see Figure 1) in active pharmaceutical ingredients (API).1,2 Although the presence of N-nitrosamines came as a huge surprise to industry and regulators alike, both sides rapidly responded and strategies were introduced to address the problem.3

Figure 1: Structures of NDMA (ChemSpider Reference 5894) and NDEA (ChemSpider reference 5708).
However, it soon became clear that this wasn’t just an API contamination issue. First, some of the H-2 blockers, ie, ranitidine, nizatidine (see Figure 2), were shown to produce NDMA on stability storage.4
This thermal degradation can also occur artefactually during analysis, ie, in the gas chromatography injection port. The US Food and Drug Administration (FDA) subsequently developed a liquid chromatography – high resolution mass spectroscopy (LC-HRMS) method for the determination of NDMA in ranitidine.1,2
The second occurrence of NDMA arising from degradation was in metformin drug product.5 The root cause of the problem was the reaction of dimethylamine (DMA), an impurity in the API (Ph. Eur., impurity F), with nitrites occurring as biproducts in the formulation excipients.6 Under acidic conditions nitrites can nitrosate the DMA to produce NDMA. Elevated levels also arose due to analytical interference.
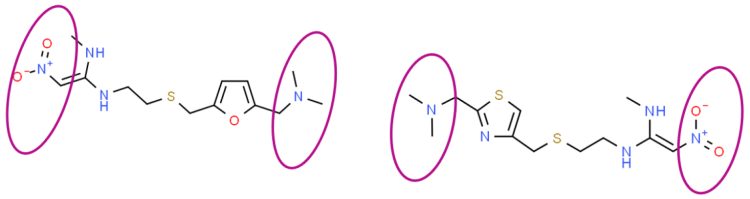
Figure 2: Structures of ranitidine (ChemSpider Reference 2272523, LHS), nizatidine (ChemSpider Reference 2298266, RHS) highlighting that intra-molecular re-arrangement can generate NDMA.
Dimethylformamide (DMF) – a common residual solvent in metformin HCl – has a molecular weight (MW) of 73.095 g/mol and NDMA has a MW of 74.0819 g/mol. Therefore, unless HRMS is used for the analysis of samples the presence of DMF interferes in the accurate mass determination of NDMA, resulting in artefactually elevated levels of NDMA.7
A recent comparative study of highly sensitive NDMA analytical methods in various metformin drug products, across four different laboratories, evaluated 2 x LC-MS/MS, GC–MS/MS and GC‑HRMS methods. The study found good agreement for the instant release (IR) metformin formulations. However, higher levels of NDMA were found in the extended release (XR) metformin formulation using GC-MS/MS compared to LC-MS/MS due to artefactual formation of NDMA.8
Only recently has the full extent of residual nitrite contamination in excipients become apparent.6 The control strategy for metformin XR products involved:
(i) double recrystallisation of metformin HCl to reduce residual levels of DMA
(ii) selection of polyvinylpyrrolidone (PVP) and hydroxypropyl methylcellulose (HPMC) supplies with reduced levels of residual nitrite.5
The third, and by far the most significant, occurrence of degradation resulting in N-nitrosamines is the formation of API-nitrosamines or nitrosamine drug substance-related impurities (NDSRI).9,10 Here the API, which is typically a secondary amine, reacts with nitrites in excipients6 under acidic conditions to form the NDSRI.11
These NDSRIs would be expected to be far less mutagenic than the small alkyl N-nitrosamines, ie, NDMA or NDEA, as increased molecular size, weight, electronic and steric factors tend to deactivate these larger N-nitrosamines.12-16
N-nitrosamines tend to be very heterogenous in nature, with nearly 20 percent non-mutagenic, eg, N-nitrosovalsartan.16,17 Against this background, it was always difficult to understand why regulatory agencies were keen to assign very low acceptable intakes (AI) for new NDSRIs.
FDA assigned a provisional AI of 26.5 ng/day; while the EMA assigned a provisional AI of 18 ng/day for new NDSRIs. However, EMA has subsequently introduced a temporary AI (t‑AI) of 178 ng/day until a substance‑specific AI can be assigned. This t-AI would be used for ≤12 months.10 It is not clear whether FDA will adopt a similar t-AI.
Regulatory guidance on acceptable intakes
There is no clear guidance from regulatory agencies on how to assign a whole life AI based on read across approaches”
There is no clear guidance from regulatory agencies on how to assign a whole life AI based on read across approaches, which make use of compound-specific data for N-nitrosamines with a similar structure.1,2 Of the limited number of N-nitrosamines in the Lhasa Cancer Potency Database (LCPD), less than four percent are NDSRIs.15 Consequently, only that portion of the NDSRI that is adjacent to the N-nitrosamine moiety can be used for read-across approaches, plus the reference mutagen will be a small MW N-nitrosamine. This artificially reduces the whole life AI of the NDSRI, as the reference mutagen is smaller in size and more potent than the corresponding NDSRI.
In the case of N-nitroso varenicline (NNV) the most obvious small MW reference mutagen would be N-nitrospiperidine (AI of 1,300 ng/day), forming the bridged part of the bicyclic seven-membered ring; but regulatory agencies selected N-nitroso‑1,2,3,6‑tetrahydropyridine as the reference mutagen (AI of 37 ng/day). However, the bridged bicyclical ring of NNV is saturated, ie, no double bond character and the introduction of a double bond into the reference mutagen substantially increases the potency of the resultant mutagen.18
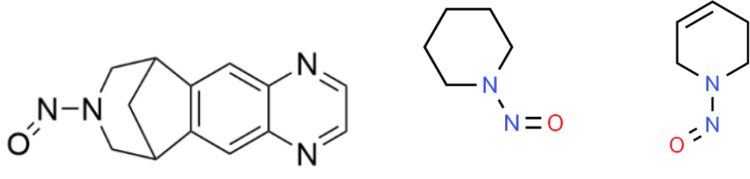
Figure 3: Structure of N-nitrosovarenicline (NNV), N-nitrosopiperidine (ChemSpider Reference 7245) and N-nitroso 1,2,3,6-tetrahydropyridine (ChemSpider Reference 37838).
And additionally, the Ames test profile of NNV, ie, positive in Salmonella strain TA153518 with metabolic activation (+S9), is not indicative of a potent mutagen, ie, an AI of 37 ng/day. Instead, the short chain alkyl N-nitrosamines, eg, NDEA (AI 26.5 ng/day) are positive in two Salmonella strains, ie, (TA1535 and TA100) and E. coli WP2 uvrA.19 This would strongly suggest that the allocated AI for NNV is significantly over-estimated. The other concerning factor is that the regulatory agencies refuse to accept that a negative Ames test discharges the risk of mutagenicity.9,10
Regulatory agencies have mandated that additional in vitro/in vivo tests must be applied to support the negative Ames test findings.”
This is contrary to ICH M7 guidance.20 This decision is based on historical data of doubtful providence that show that the very potent, short alkyl chain nitrosamines affect the metabolic activation of these mutagens and make them appear less potent. Regulatory agencies have mandated that additional in vitro/in vivo tests must be applied to support the negative Ames test findings. Glowienke et al.17 recently published data for a valsartan-related complex N-nitroso-impurity, nitrosated intermediate 181-14, highlighting that the negative Ames finding was indeed confirmed by a negative transgenic gene mutation assay. The authors indicated that the Ames test was still a valid assessment of mutagenicity.
Based on our structural understanding of the metabolic activation of N-nitrosamines, this intermediate 181-14 would not be expected to be mutagenic due to the presence of a bulky constituent on the alpha-carbon. Trejo-Martin et al.21 also showed that an Ames test using pre-incubation protocol in dimethyl sulfoxide (DMSO) with either rat or hamster S9 for metabolic activation could demonstrate the potential mutagenicity of N-nitrosamine mutagens.
ICH M720 indicates that, “the use of the appropriate acceptable intake can be justified in cases of severe disease, reduced life expectancy, late onset but chronic disease, or with limited therapeutic alternatives”. ICH S922 applies for cancer treatments and the limits are aligned with ICH Q3A or Q3B. This is also applicable to N-nitrosamine impurities.9,10
Establishing sensitive analytical methods
The availability of a suitable specific and highly sensitive method for these newly identified NDSRIs is a significant challenge”
The availability of a suitable specific and highly sensitive method for these newly identified NDSRIs is also a significant challenge.1,2 The most sensitive method is US Pharmacopeia (USP) procedure 4 (GC‑MS/MS – triple quad), which has a limit of quantitation (LoQ) of 5ppb.23 This has been validated but only in API matrices. In drug products the presence of excipients means the method would struggle to achieve 5ppb LoQ without additional extraction/enrichment strategies. For drug products with a maximum daily dose (MDD) of > 2,000 mg/day and with an AI of 18 ng/day,10 the concentration limit is 0.009ppm or 9ppb (18/2,000). EMA requires that the limit should be < 10 percent of AI, which equates to 0.9ppb.
Consequently, USP procedure 4 would not have the required sensitivity. EMA has indicated that, “Exceptions are anticipated for medicinal products used at high daily dose (AI may be below the technical feasibility of the method), or in cases where more than one nitrosamine is anticipated or identified in given medicinal product.”10
European actions on NDSRIs: MutaMind project
The regulatory agencies are aware of the many issues that bedevil assessment of NDSRIs. In the EU, the EMA-MutaMind project has been initiated to try and address these problems.24
There are four main streams that cover the following:
(i) Formation of NDSRIs endogenously from the reaction of API and nitrite under realistic conditions in different parts of the gastrointestinal tract. ICH M720 indicates that, “Higher acceptable intakes may be justified when human exposure to the impurity will be much greater from other sources eg, food, or endogenous metabolism.”
(ii) Metabolic activation of NDSRIs and identification of the enzyme systems involved
(iii) Types of DNA adducts, kinetics and repair mechanisms
(iv) Optimisation of in vitro mutagenicity assays, eg, the Ames test.
In conclusion, AIs and control strategies for short chain alkyl N-nitrosamines, ie, NDMA in APIs, were relatively easily established. In contrast, NDSRIs have proved to be highly challenging, as there are no meaningful reference mutagens that adequately reflect their molecular size and complexity. Using small molecule reference mutagens generates artefactually low AIs and oftentimes the methods are inadequately sensitive to accurately quantitate and control the NDSRIs. The Ames test, the cornerstone of mutagenic toxicology, has been challenged by regulators even though the available information, albeit limited, demonstrates that negative Ames tests are validated by other in vivo mutagenic methods, ie, MutaMice. However, potentially the biggest challenge is how to justify ppb limits for NDSRIs if these same molecules are endogenously produced in the body at percentage levels.
About the author
Dave Elder, PhD has worked in the pharmaceutical industry for 45 years for GSK, Syntex and Sterling. He is currently a CMC consultant with an interest in impurities and safety‑based limits. Dave is a member of the British Pharmacopoeia Expert Advisory Group, PCN (Pharmacy and Nomenclature) and a former member of RSC’s Analytical Division Council and JPAG (Joint Pharmaceutical Analysis Group).
References
1. Elder DP. Johnson GE, Snodin DJ. Tolerability of risk: A commentary on the nitrosamine contamination issue. J Pharm Sci. 2021; 110(6): 2311-2328.
2. Snodin DJ, Elder DP. Short commentary on NDMA (N-nitrosodimethylamine) contamination of valsartan products. Reg Toxicol Pharmacol. 2019; 103: 325–329.
3. EMA [Internet]. Lessons learnt from presence of N-nitrosamine impurities in sartan medicines. 2020 [cited 2023Jan]. Available from: https://www.ema.europa.eu/en/documents/report/lessons-learnt-presence-n-nitrosamine-impurities-sartan-medicines_en.pdf
4. King FJ, Searle AD, Urquhart MW. Ranitidine—Investigations into the Root Cause for the Presence of N-Nitroso-N,N-dimethylamine in Ranitidine Hydrochloride Drug Substances and Associated Drug Products. J Pharm Sci. 2020; 24(12): 2915-2926.
5. Schlingemann J, Boucley C, Hickert S, et al. Avoiding N-nitrosodimethylamine formation in metformin pharmaceuticals by limiting dimethylamine and nitrite. Int J Pharm. 2022; 620: 121740.
6. Boetzel R, Schlingemann J, Hickert S, et al. A Nitrite Excipient Database: A useful Tool to Support N-Nitrosamine Risk Assessments for Drug Products. J Pharm Sci. 2022; S0022-3549(22)00168-X.
7. Yang J, Marzan TA, Ye W, et al. A cautionary tale: Quantitative LC-HMRS analytical procedures for the analysis of N-nitrosodimethylamine in metformin. AAPS J. 2020; 22(4): 89.
8. Fritzsche M, Blom G, Keitel J, et al. NDMA analytics in metformin products: Comparison of methods and Pitfalls. Eur J Pharm Sci. 2022; 168: 106026.
9. FDA. Updates on possible mitigation strategies to reduce the risk of nitrosamine drug substance-related impurities in drug products [Internet]. FDA, Center for Drug Evaluation and Research; 2021 [cited 2023Jan]. Available from: https://www.fda.gov/drugs/drug-safety-and-availability/updates-possible-mitigation-strategies-reduce-risk-nitrosamine-drug-substance-related-impurities
10. EMA. Questions and Answers for marketing authorisation holders/applicants on the CHMP Opinion for the Article 5(3) of Regulation (EC) No 726/2004 referral on nitrosamine impurities in human medicinal products. EMA/409815/2020 Rev.13.
11. López-Rodríguez R, McManus JA, Murphy NS, et al. Pathways for N-Nitroso Compound Formation: Secondary Amines and Beyond. Org Proc Res Dev. 2020; 24(9): 1558–1585.
12. Cross KP, Ponting DJ. Developing structure-activity relationships for N-nitrosamine activity. Comput Toxicol. 2021; 20: 100186.
13. Thomas R, Thresher A, Ponting DJ. Utilisation of parametric methods to improve percentile-based estimates for the carcinogenic potency of nitrosamines. Regul Toxicol Pharmacol. 2021; 121: 104875.
14. Thomas R, Tennant RE, Oliveira AA, et al. What Makes a Potent Nitrosamine? Statistical Validation of Expert-Derived Structure–Activity Relationships. Chem Res Toxicol. 2022; 35(11): 1997–2013.
15. Schlingemann J, Burns MJ, Ponting DJ, et al. The landscape of potential small and drug substance related nitrosamines in pharmaceuticals. J Pharm Sci. 2022.
16. Thresher A, Foster R, Ponting DJ, et al. Are all nitrosamines concerning? A review of mutagenicity and carcinogenicity data. Regul Toxicol Pharmacol. 2020; 116: 104749.
17. Glowienke S, Onken U, Elhajouji A, et al. Genotoxicity evaluation of a valsartan-related complex N-nitroso-impurity. Reg Toxicol Pharmacol. 134; 2022: 105245.
18. Ponting DJ, Dobo KL, Kenyon M Kalgutkar AS. On the carcinogenic potential of novel N-nitrosamine impurities derived from active pharmaceutical ingredients. J Med. Chem. 2022; 65(23): 15584–15607.
19. Bringezu F, Simon S. Salmonella typhimurium TA100 and TA1535 and E. coli WP2 uvrA are highly sensitive to detect the mutagenicity of short Alkyl-N-Nitrosamines in the Bacterial Reverse Mutation Test. Toxicol Rep. 2022; 9: 250–255.
20. ICH M7 Guideline on Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk. [Internet] EMA. 2014. [cited 2023Jan] Available from: https://www.ema.europa.eu/en/ich-m7-assessment-control-dna-reactive-mutagenic-impurities-pharmaceuticals-limit-potential
21. Trejo-Martin A, Bercu JP, Thresher A, et al. Use of the bacterial reverse mutation assay to predict carcinogenicity of N-nitrosamines. Regul Toxicol Pharmacol. 135; 2022: 105247.
22. ICH S9 Guideline: Nonclinical Evaluation for Anticancer Pharmaceuticals. ICH. May 2010. EMA/CHMP/ICH/646107/2008. [Internet]. EMA. 2022. [cited 2023Jan]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-s-9-nonclinical-evaluation-anticancer-pharmaceuticals-note-guidance-nonclinical-evaluation_en.pdf
23. Biba E. GENERAL CHAPTER <1469> NITROSAMINE IMPURITIES. Stakeholder Forum November 19, 2020. [Internet] [cited 2023Jan]. Available from: https://www.usp.org/sites/default/files/usp/document/stakeholder-forum/pnp/highlights-of-1469-nitrosamine-impurities.pdf.
24. Cross KP, Bassan A, Brandsma I, et al. N-nitrosamine impurities in drugs, introducing EMA-Mutamind project. [Internet] Instem. 03 November 2022. [cited 2023Jan]. Available from: https://www.instem.com/industries/video/introducing-the-ema-mutamind-project.php