New developments in the pharmaceutical stress testing industry
Posted: 22 December 2021 | Ana Cláudia Santos (Merck Brazil), Chris Foti (Gilead Sciences), Flavia Firmino (Pfizer), John Campbell (GlaxoSmithKline), Steven Baertschi (Baertschi Consulting) | No comments yet
A team of American scientists report on some recent developments in the realm of pharmaceutical stress testing, or forced degradation, practices relating to solid dosage forms, bringing some valuable clarity for drug developers.
Introduction
Stress testing, or forced degradation, has long been recognised by the pharmaceutical industry as an important part of the drug development process.1-5 Legislation6 and associated guidelines7,8 from the Brazilian regulatory authority (Agência Nacional de Vigilância Sanitária, or ANVISA) have introduced detailed regulatory requirements for forced degradation studies, including many requirements that are globally unique. For example, one requirement of RDC 53/2015 is that solution phase stress testing be conducted not only for drug substances, but also for drug products.
These are burdensome experiments to perform, since the design of the study is more complicated when the solid dosage form is exposed to solutions of low pH, high pH, oxidants and metal ions. Difficulties with extraction and dissolution of the active pharmaceutical ingredient (API) from the dosage form are more common with solid dosage forms, and such difficulties can lead to lack of mass balance and other complications. Additionally, providing justification for not performing these experiments has not always been accepted by ANVISA and pharmaceutical companies have had limited success in regulatory submissions without meeting these requirements.
To address these issues, a benchmarking study was initiated by a cohort of scientists from nine pharma companies and representation from ANVISA with the goal of evaluating the scientific merits of solution-phase stress testing of solid drug products within the context of stress testing best practices across the pharmaceutical industry and health authorities. The results of this effort have been published recently in the Journal of Pharmaceutical Sciences.9
Regulatory overview of forced degradation
The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) discuss stress testing in many of their guidelines.10-13 The subject is also addressed in agency-specific guidelines available from the US Food and Drug Administration (FDA),14 World Health Organization (WHO)15 and European Medicines Agency (EMA).16 Global regulatory expectations for stress testing studies have also been reviewed in literature.5
 Despite obvious regulatory expectations, the specifics of stress testing study designs are not explicitly prescribed in most regulatory guidelines. This is because most studies must be customised based on the properties of the drug substance and/or drug product under examination. For this reason, the details of stress testing experiments are generally left to the applicant.
Despite obvious regulatory expectations, the specifics of stress testing study designs are not explicitly prescribed in most regulatory guidelines. This is because most studies must be customised based on the properties of the drug substance and/or drug product under examination. For this reason, the details of stress testing experiments are generally left to the applicant.
However, Brazilian regulatory expectations since 2015 have been both globally unique and prescriptive, thus attracting much attention from the pharmaceutical industry. Since the enaction of RDC 53/2015, Brazil joined ICH in 2016 and is now a participant in global efforts to harmonise pharmaceutical regulatory practice. As a result of these developments, there is substantial interest in scientific dialogue between ANVISA and industry partners, resulting in a collaboration to assess the utility of the stress conditions prescribed in Brazilian legislation.
What did the benchmarking study show?
The goal of the study was to determine the extent to which solution-phase stress testing of solid drug products generates unique degradation products not observed under any other condition of a well‑designed stress testing study, then determine if any of those unique degradation products were relevant to the long-term stability data. Venn diagrams were used to compare the stability data from 62 drug products and their corresponding stress testing results from ANVISA and participating pharmaceutical companies in this benchmarking study.
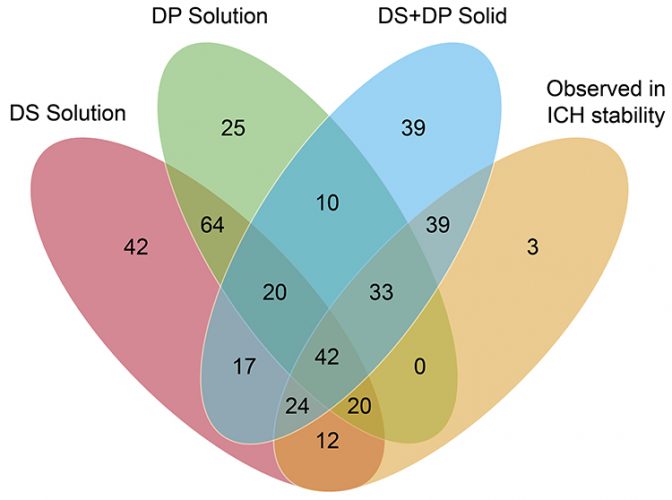
Figure 1: Venn diagram illustrating distribution of degradation products in the benchmarking study.
A review of the data showed that 387 potential degradation products were reported to have been observed during the stress testing experiments conducted for the 62 drug products, with only 173 actual degradation products observed to form during accelerated or long-term storage conditions above the reporting threshold stipulated in ICH Q3B(R2).9 These “actual” degradation products were therefore considered “relevant.” It is clear from Figure 1 that solution-phase stress testing of the drug products yielded no degradation products that were both unique and relevant to the stability data. This point is illustrated more clearly in Figure 2, which plots only the 173 relevant degradation products, categorised according to the stress conditions that induced their formation. There were also three degradation products that were not observed in any of the other stress testing conditions. This was attributed to the fact that these degradation products were observed only under the final packaged conditions containing desiccant and, therefore, would not have been observed in traditionally designed stress testing studies.
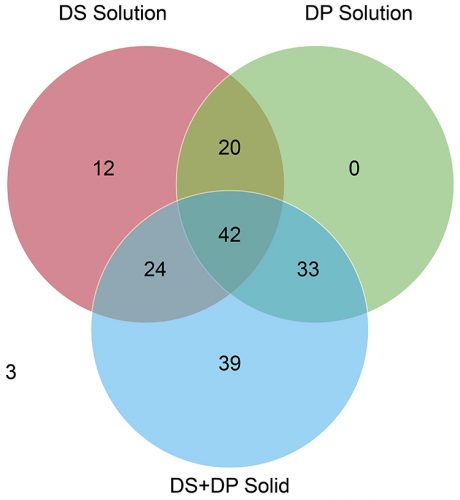
Figure 2: Venn diagram illustrating distribution of degradation products from stress testing studies observed during ICH stability studies.
Submission implications
The results of the benchmarking study presented in this review (and in more detail in the Journal of Pharmaceutical Sciences)9 were presented to ANVISA17 and Sindicato Da Indústria De Produtos Farmacêuticos No Estado De São Paulo (Sindusfarma).18 This provided the agency the scientific basis to plan a future review of the RDC 53. As a result, the revision of RDC 53/2015 has been included in the ANVISA’s Regulatory Agenda (2021-2023) and the ANVISA’s Technical Area (GQMED). ANVISA indicated during the Sindusfarma meeting in June 2021 their intention to review the regulation. The next steps from ANVISA to update the regulation are:
- The initiation of the official regulatory process to review the RDC 53
- Updating the technical text
- Public consultation publication.
The final review of the regulation is expected to occur in 2022.
Stress testing, or forced degradation, has long been recognised by the pharmaceutical industry as an important part of the drug development process”
The findings and conclusions of this benchmarking study are considered by ANVISA as a robust technical reference. Since the current regulation allows for providing justification for not performing some of the defined experiments, companies can refer to this publication to justify exclusion of the solution-phase stress testing of solid drug products, even while the review of the current ANVISA’s regulation remains in progress. It should be noted that, in some cases, additional information regarding the quality of the method and its stability-indicating capability may be necessary.
In addition to these developments, prior to revision of the regulation ANVISA intends to update the current Q&A document associated with RDC 53/2015 to formalise the acceptance of the justifications based on this benchmarking study.
The recommended best practice for conducting stress testing studies is shown in Figure 3. Although this cross-industry effort was mainly focused on Brazilian requirements, the results may be applied to stress testing globally, including by other regulators worldwide. The benchmarking study not only outlines the results of a wide range of products obtained by a large number of pharmaceutical companies, but also makes recommendations that will improve the consistency of international science-driven practices.
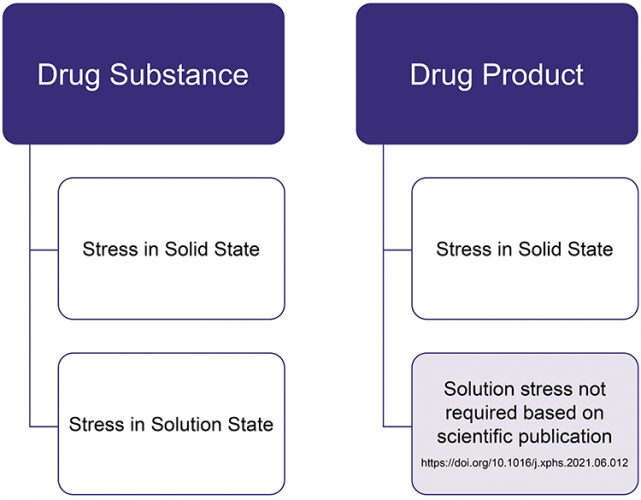
Figure 3: Stress testing design for solid dosage forms.
Future efforts for stress testing
The success of the benchmarking study has led ANVISA to express interest in further collaborative efforts on additional topics pertaining to pharmaceutical stress testing studies, especially in light of the planned revision of RDC 53/2015 and alignment with ICH. Such additional topics of interest include best practices for evaluating mass balance19-21 in forced degradation samples, as well as scientific approaches for determining whether sufficient stress has been applied when minimal degradation is observed in stress testing experiments.
Future collaborative efforts will hopefully bring additional clarity to these and other topics as regulators and industry attempt to tackle some of the more challenging aspects of the “artful science”22 of pharmaceutical stress testing.
About the authors
 Dr Steven Baertschi is President of Baertschi Consulting, a firm specialising in solving difficult stability, impurity, analytical, solid-state and formulation issues. He obtained his PhD in Organic Chemistry (1989) from Vanderbilt University and was named a Fellow of the AAPS in 2007. Retiring from Eli Lilly & Co after 26 yrs, Dr Baertschi has chaired numerous scientific conferences on stress testing, [photo] stability and impurities, and has published more than 90 scientific articles / book chapters, including two editions of a book on drug degradation.
Dr Steven Baertschi is President of Baertschi Consulting, a firm specialising in solving difficult stability, impurity, analytical, solid-state and formulation issues. He obtained his PhD in Organic Chemistry (1989) from Vanderbilt University and was named a Fellow of the AAPS in 2007. Retiring from Eli Lilly & Co after 26 yrs, Dr Baertschi has chaired numerous scientific conferences on stress testing, [photo] stability and impurities, and has published more than 90 scientific articles / book chapters, including two editions of a book on drug degradation.
 Dr Chris Foti is currently a Director in Analytical Operations at Gilead Sciences with over 20 years of experience in the pharmaceutical industry in Analytical CMC. He has worked at Abbott Laboratories and Pfizer during his career and received his PhD in Organic Chemistry from North Carolina State University followed by postdoctoral research at the University of North Carolina at Chapel Hill.
Dr Chris Foti is currently a Director in Analytical Operations at Gilead Sciences with over 20 years of experience in the pharmaceutical industry in Analytical CMC. He has worked at Abbott Laboratories and Pfizer during his career and received his PhD in Organic Chemistry from North Carolina State University followed by postdoctoral research at the University of North Carolina at Chapel Hill.
 John Campbell is a Scientific Leader and GSK Fellow at GlaxoSmithKline in the greater Philadelphia area. He specialises in the analytical characterisation of pharmaceuticals in clinical development with special expertise in degradation chemistry and stress testing methodology for both small molecules and therapeutic proteins. He holds a PhD in organic chemistry from Washington University in St. Louis.
John Campbell is a Scientific Leader and GSK Fellow at GlaxoSmithKline in the greater Philadelphia area. He specialises in the analytical characterisation of pharmaceuticals in clinical development with special expertise in degradation chemistry and stress testing methodology for both small molecules and therapeutic proteins. He holds a PhD in organic chemistry from Washington University in St. Louis.
 Flavia Firmino is the Regional Regulatory Advisor for Latin America region in Global CMC at Pfizer. She holds accountability for partnering with manufacturing organisations and Regulatory Affairs while providing regional regulatory expertise to CMC Product Strategists, addressing strategic issues and helping to develop policies and positions on draft regulations in regional and external engagement on CMC topics. She has 20 years’ experience in the pharma industry in Quality and Regulatory areas and held leadership positions in Regulatory Conformance, Quality Operations and Regulatory Affairs (country level). Flavia holds a degree in Pharmacy & Biochemistry from the Oswaldo Cruz University in Brazil and a post graduate degree in Industrial Administration.
Flavia Firmino is the Regional Regulatory Advisor for Latin America region in Global CMC at Pfizer. She holds accountability for partnering with manufacturing organisations and Regulatory Affairs while providing regional regulatory expertise to CMC Product Strategists, addressing strategic issues and helping to develop policies and positions on draft regulations in regional and external engagement on CMC topics. She has 20 years’ experience in the pharma industry in Quality and Regulatory areas and held leadership positions in Regulatory Conformance, Quality Operations and Regulatory Affairs (country level). Flavia holds a degree in Pharmacy & Biochemistry from the Oswaldo Cruz University in Brazil and a post graduate degree in Industrial Administration.
 Ana Cláudia Santos is a Global Analytical Technology Lead Expert at Merck Brazil. With over 18 years of experience in the pharmaceutical industry, Ana’s background covers diversified technical know-how and leadership positions within Analytical R&D, Industrial Development, CMC Regulatory Compliance, and Quality Systems. Prior to Merck, she worked at Pfizer, Sanofi and Medley. Ana holds a degree in Pharmacy & Biochemistry, MBA in Business Management and postgraduate certificates in Analytical Sciences and Statistics.
Ana Cláudia Santos is a Global Analytical Technology Lead Expert at Merck Brazil. With over 18 years of experience in the pharmaceutical industry, Ana’s background covers diversified technical know-how and leadership positions within Analytical R&D, Industrial Development, CMC Regulatory Compliance, and Quality Systems. Prior to Merck, she worked at Pfizer, Sanofi and Medley. Ana holds a degree in Pharmacy & Biochemistry, MBA in Business Management and postgraduate certificates in Analytical Sciences and Statistics.
Acknowledgements
The authors acknowledge the following individuals across the pharmaceutical industry and ANVISA for their contributions to the Benchmarking Study Publication in the Journal of Pharmaceutical Sciences: Chloe Wang, Neal Adams, Leonardo R. Allain, Gabriela Araujo, Renan Azevedo, Juçara Ribeiro Franca, Simon R Hicks, Steven Hostyn, Patrick J Jansen, Dorina Kotoni, Andreas Kuemmell, Stacey Marden, Gregory Rullo, Gregory W Sluggett and Todd Zelesky.
References
- Baertschi SW, Alsante KM, Reed RA, eds. Pharmaceutical Stress Testing: Predicting Drug Degradation, 2nd London: Informa Healthcare; 2011.
- Reynolds DW, Facchine KL, Mullaney JF, et al. Available guidance and best practices for conducting forced degradation studies. Pharm Technol 2002;26:48-56.
- Alsante KM, Ando A, Brown R, et al. The role of degradant profiling in active pharmaceutical ingredients and drug products. Adv Drug Deliv Rev 2007;59:29-37.
- Alsante KM, Baertschi SW, Coutant M, et al. Degradation and Impurity Analysis for Pharmaceutical Drug Candidates. In: Ahuja S, Scypinski S, eds. Handbook of Modern Pharmaceutical Analysis, 2nd Vol 10 of Separation Science and Technology. Elsevier;2010:59-169.
- Singh S, Junwal M, Modhe G, et al. Forced degradation studies to assess the stability of drugs and products. TrAC Trends in Analytical Chemistry 2013;49:71-88.
- Brazilian Ministry of Health, National Health Surveillance Agency (ANVISA). Establishes parameters for reporting, identification and qualification of degradation products in drug products with synthetic and semi-synthetic active substances, classified as new chemical entities, generics and branded generics, among other provisions. Resolution RDC 53/2015
- National Health Surveillance Agency (ANVISA). Guide for obtaining degradation profile, and identification and qualification of degradation products in drug products. Guide No. 04/2015.
- National Health Surveillance Agency (ANVISA). Questions & Answers, Subject: RDC 53/2015 and Guide 04/2015. Version 1.3, 2017.
- Campbell JM, Foti C, Wang C, et al. “Assessing the Relevance of Solution Phase Stress Testing of Solid Dosage Form Drug Products: A Cross-Industry Benchmarking Study.” Pharm. Sci. 2021, In Press (doi: 10.1016/j.xphs.2021.06.012)
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), Stability Testing of New Drug Substances and Products, Q1A(R2), 2003.
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Validation of Analytical Procedures: Text and Methodology. Q2(R1), 2005.
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Impurities in New Drug Substances. Q3A(R2), 2006.
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Impurities in New Drug Products. Q3B(R2), 2006.
- United States Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER). INDs for Phase 2 and Phase 3 Studies: Chemistry, Manufacturing, and Controls. Guidance for Industry, docket number FDA-1999-D-0030, 2003.
- World Health Organization (WHO) Expert Committee on Specifications for Pharmaceutical Preparations. Stability testing of active pharmaceutical ingredients and finished pharmaceutical products. WHO 52nd Technical Report Series, No. 1010, 2018, pp 309-351.
- European Medicines Agency (EMEA), Committee for Proprietary Medicinal Products (CPMP). Guideline on the Requirements to the Chemical and Pharmaceutical Quality Documentation Concerning Investigational Medicinal Products in Clinical Trials. CHMP/QWP/185401/2004 final. London, UK, 2006.
- Rullo G, Baertschi SW, Campbell JM, Adams N. “Assessing the relevance of solution phase stress testing of solid dosage form drug products: a cross-industry benchmarking study.” Virtual Workshop with ANVISA. April 2021. (oral presentation)
- Fermino F, Hicks S, Foti C, et al. “Assessing the relevance of solution phase stress testing of solid dosage form drug products: a cross-industry benchmarking study.” Virtual Workshop with Sindusfarma. 28 April 2021. (oral presentation)
- Nussbaum MA, Kaerner A, Jansen PJ, Baertschi SW, “Role of ‘mass balance’ in pharmaceutical stress testing”, in , Pharmaceutical Stress Testing: Predicting Drug Degradation, 2nd edition, Baertschi SW, Alsante, KM, Reed RA, Eds, Informa Healthcare, London (2011).
- Baertschi SW, “Analytical Methodologies for Discovering and Profiling Degradation-Related Impurities”, Trends in Analytical Chemistry, 25:8, 758-767 (2006).
- Baertschi SW, Pack BW, Hoaglund-Hyzer CS, Nussbaum MA. “Assessing Mass Balance in Pharmaceutical Drug Products: New Insights into an Old Topic”, Trends in Analytical Chemistry, 49, 126-136 (2013).
- Preface, Pharmaceutical Stress Testing: Predicting Drug Degradation, 2nd edition, Baertschi SW, Alsante, KM, Reed RA, Eds, Informa Healthcare, London (2011).
Issue
Related topics
Active Pharmaceutical Ingredient (API), Analytical techniques, Drug Development, Drug Safety, Formulation, ICH guidelines, Industry Insight, Regulation & Legislation, Therapeutics
Related organisations
Agência Nacional de Vigilância Sanitária (ANVISA), International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH), Sindicato Da Indústria De Produtos Farmacêuticos No Estado De São Paulo (Sindusfarma), The European Medicines Agency (EMA), The World Health Organization (WHO), US Food and Drug Administration (FDA)